Credito:Università della Carolina del Nord presso la Chapel Hill School of Medicine
Gli scienziati della UNC School of Medicine in collaborazione con i ricercatori dell'Oregon Health &Science University hanno risolto la struttura molecolare tridimensionale della proteina che è difettosa nelle persone con fibrosi cistica nello stato attivo e inattivo della proteina. La scoperta, pubblicato sulla rivista Biochimica , potrebbe aprire nuove strade di ricerca e aiutare gli sviluppatori di farmaci a creare farmacoterapie avanzate per aiutare le persone con fibrosi cistica.
Gran parte del lavoro di biochimica è stato svolto nel laboratorio di John Riordan, dottorato di ricerca, illustre professore di biochimica e biofisica all'UNC-Chapel Hill. Alla fine degli anni '80, Il laboratorio di Riordan ha scoperto il gene mutato responsabile della fibrosi cistica. Se un bambino riceve una copia di questo gene difettoso da ciascun genitore, il bambino svilupperà CF. La proteina codificata da questo gene è stata definita il regolatore transmembrana della fibrosi cistica, o CFTR, che è il canale del cloro nelle cellule epiteliali che popolano il sistema respiratorio. Le persone con FC mancano di un canale epiteliale del cloro funzionale, che è essenziale per mantenere il corretto equilibrio di sale e acqua nei polmoni e in altri organi. Un risultato di ciò è la produzione di spessi, muco appiccicoso che diventa difficile da rimuovere dalle vie aeree, portando a infezioni croniche e una durata di vita più breve per la maggior parte delle persone con FC.
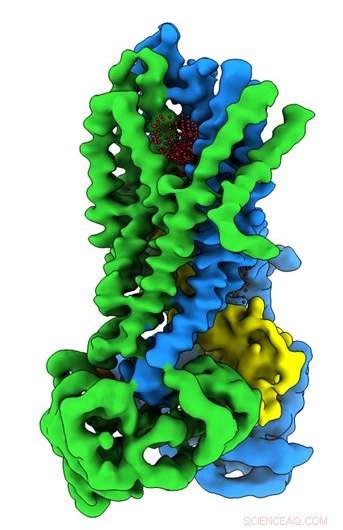
Nel laboratorio Riordan, borsista postdottorato Jonathan Fay, dottorato di ricerca, ha condotto esperimenti utilizzando la microscopia crioelettronica a particella singola per scoprire la struttura molecolare del CFTR in presenza di ATP, una sostanza chimica organica complessa necessaria per molti processi nelle cellule, compreso un canale ionico cloruro funzionante cruciale per la corretta funzione polmonare. Per aiutare a catturare le strutture della proteina CFTR nel suo stato attivo e inattivo, il laboratorio Riordan ha stabilizzato la proteina CFTR in modo tale che il canale fosse spento quando defosforilato e bloccato quando fosforilato.
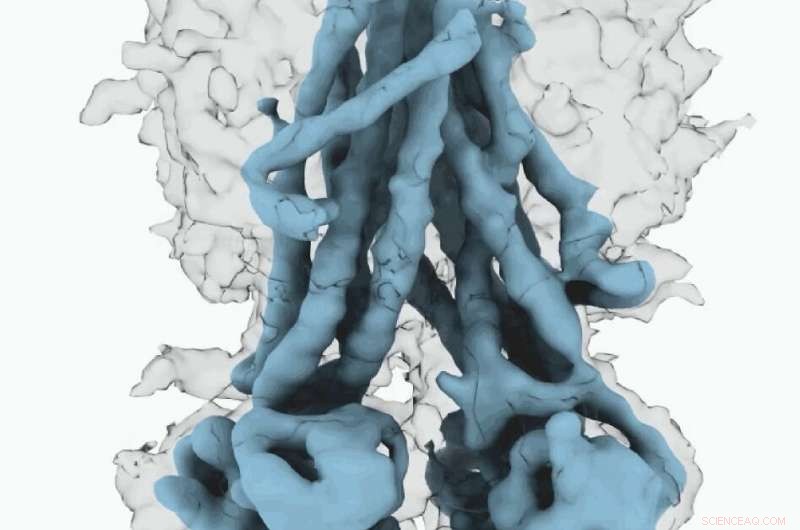
Mappa Cryo-EM di CFTR. Credito:Università della Carolina del Nord presso la Chapel Hill School of Medicine
Queste strutture molecolari rivelano un riposizionamento unico di parti della proteina CFTR, fornendo approfondimenti sulla transizione strutturale tra stati funzionali attivi e inattivi di CFTR.
Inoltre, Fay e colleghi hanno osservato dettagli di questo complesso proteico che differiscono da ciò che altri scienziati hanno scoperto nelle precedenti strutture CFTR.
"È davvero sorprendente quanto le tecnologie crio-EM siano avanzate e come l'utilizzo di queste tecniche possa permetterci di visualizzare diversi stati del canale, " Ha detto Fay. "Penso che i nostri risultati siano molto entusiasmanti. Abbiamo scoperto un nuovo portale che presenta una nuova promettente area del canale per indirizzare e controllare la funzione del canale CFTR."
E se i ricercatori possono mirare e controllare con successo la funzione di quel canale, quindi potrebbero creare terapie più precise per trattare meglio alcune persone con FC.