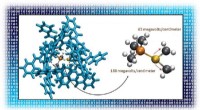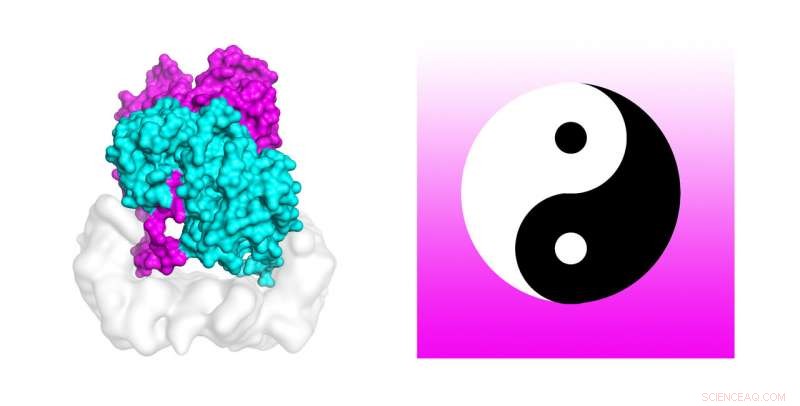
Gli scienziati stanno utilizzando potenti supercomputer per scoprire il meccanismo che attiva le mutazioni cellulari presenti in circa il 50 percento dei melanomi. Le simulazioni di dinamica molecolare sul supercomputer Stampede2 di TACC hanno testato la stabilità della struttura del complesso B-Raf:14-3-3, che quando mutato è legato al cancro della pelle. Gli autori dello studio confrontano il dimero B-Raf con il simbolo circolare cinese yin-yang di opposti interconnessi uniti alla coda. Credito:Karandur et al., TACC
Inizia in piccolo, solo un difetto della pelle. I nei più comuni rimangono così:ammassi innocui di cellule della pelle chiamate melanociti, che ci danno pigmento. In rari casi, ciò che inizia come un neo può trasformarsi in melanoma, il tipo più grave di cancro della pelle umana perché può diffondersi in tutto il corpo.
Gli scienziati stanno utilizzando potenti supercomputer per scoprire il meccanismo che attiva le mutazioni cellulari presenti in circa il 50 percento dei melanomi. Gli scienziati affermano di sperare che il loro studio possa contribuire a una migliore comprensione del cancro della pelle e alla progettazione di farmaci migliori.
Nel 2002, gli scienziati hanno scoperto un legame tra il cancro della pelle e le mutazioni della chinasi B-Raf (Fibrosarcoma rapidamente accelerato), una proteina che fa parte della catena del segnale che inizia all'esterno della cellula e va all'interno per dirigere la crescita cellulare. Questa via del segnale, chiamata via della chinasi Ras/Raf/Mek/Erk, è importante per la ricerca sul cancro, che cerca di capire la crescita cellulare fuori controllo. Secondo lo studio, circa il 50 percento dei melanomi ha una singola mutazione specifica su B-Raf, noto come residuo di valina 600 del glutammato (V600E).
B-Raf V600E divenne così un importante bersaglio farmacologico, e negli anni successivi furono sviluppati inibitori specifici del mutante. I farmaci hanno inibito il mutante, ma è successo qualcosa di strano. Paradossalmente, calmare il mutante ha avuto un lato negativo. Ha attivato l'immutato, chinasi proteiche B-Raf di tipo selvatico, che di nuovo ha innescato il melanoma.
"Con questo sfondo, abbiamo lavorato allo studio della struttura di questa importante proteina, B-Raf, "ha detto Yasushi Kondo, un ricercatore post-dottorato presso il John Kuriyan Lab dell'UC Berkeley. Kondo è il coautore di uno studio di ottobre 2019 sulla rivista Scienza che ha determinato la struttura del complesso di proteine che compongono B-Raf e ha anche scoperto come avviene la paradossale attivazione di B-Raf.
"Volevamo studiare lo stato più simile a quello nativo della proteina per capire come è regolata nelle cellule, perché la maggior parte degli studi si è concentrata sul dominio della chinasi isolato e su come i farmaci si legano al dominio della chinasi", ha detto Kondo.
La proteina B-Raf a lunghezza intera è costituita da diversi domini collegati da regioni disordinate, qualcosa di troppo ingombrante per gli scienziati da immaginare. La tecnica di Kondo consisteva nell'usare la chimica dell'intestino per creare frammenti più piccoli, quindi cucirli per ottenere la struttura completa.
"Di conseguenza, abbiamo ottenuto una forma attiva del dimero B-Raf a lunghezza intera chiamato B-Raf co-purificato con 14-3-3 dimero, una proteina dell'impalcatura legata alla coda C-terminale B-Raf fosforilata, " ha detto Kondo.
Il gruppo di Kondo ha utilizzato la microscopia crioelettronica (crio-EM) per determinare la struttura del complesso B-Raf 14-3-3, fondamentalmente congelando criogenicamente il complesso proteico, che lo manteneva in un ambiente chimicamente attivo, ambiente quasi naturale. Successivamente lo hanno fatto lampeggiare con fasci di elettroni per ottenere migliaia di "fermo immagine". Hanno setacciato il rumore di fondo e ricostruito mappe di densità tridimensionali che mostravano dettagli precedentemente sconosciuti nella forma della molecola. E per le proteine la forma segue la funzione.
Kondo ha spiegato che la struttura ha rivelato un'organizzazione asimmetrica del complesso, formato da due serie di dimeri internamente simmetrici, o coppie di molecole legate. "Proponiamo che questa disposizione inaspettata consenta l'attivazione asimmetrica del dimero B-Raf, che è un meccanismo che fornisce una spiegazione dell'origine dell'attivazione paradossale di B-Raf da parte di inibitori di piccole molecole, " ha detto Kondo.

Il supercomputer Stampede2 presso il Texas Advanced Computing Center è una risorsa assegnata dall'Extreme Science and Engineering Discovery Environment (XSEDE) finanziato dalla National Science Foundation (NSF). Credito:TACC
L'analisi dettagliata della struttura complessa asimmetrica B-Raf 14-3-3 ha mostrato un'altra caratteristica strutturale inaspettata, descritto come il segmento distale della coda, DTS in breve, di una molecola B-Raf. Kondo ha detto che la coda di uno è legata al sito attivo dell'altro, bloccando la sua attività competendo con il legame dell'ATP. La molecola B-Raf bloccata è stabilizzata nella conformazione attiva. "Abbiamo interpretato questa struttura che questa molecola B-Raf bloccata funziona come attivatore e stabilizza l'altro ricevitore B-Raf attraverso l'interfaccia del dimero, " ha detto Kondo.
Abbastanza curiosamente, gli autori confrontano il dimero B-Raf con il simbolo circolare cinese yin-yang di opposti interconnessi uniti alla coda. "Dall'osservazione del soggetto, è molto chiaro che non si è in grado di fosforilare la molecola a valle, che è necessario per la crescita cellulare. L'altra molecola è chiaramente quella che fa il lavoro. In questo insieme di due molecole, vediamo chiaramente che uno sta facendo il lavoro di supporto, e l'altro sta facendo il lavoro vero e proprio. Sembra davvero Yin e Yang in questo complesso B-Raf 14-3-3 che abbiamo risolto, " ha detto Kondo.
Sembra, anche se, può ingannare. Gli scienziati hanno utilizzato simulazioni al computer per verificare che fossero davvero su qualcosa. "Abbiamo eseguito simulazioni di dinamica molecolare di questo complesso del dimero B-Raf legato a un dimero 14-3-3 per testare la stabilità della conformazione asimmetrica, " ha detto il coautore dello studio Deepti Karandur, anche un ricercatore post-dottorato presso il John Kuriyan Lab dell'UC Berkeley; è anche una borsista post-dottorato presso l'Howard Hughes Medical Institute. "Non sapevamo perché la conformazione fosse asimmetrica, o quale ruolo ha svolto nel mantenere lo stato attivo dell'enzima, " ha detto Karandur.
Hanno iniziato le simulazioni utilizzando la struttura che Kondo aveva risolto con cryo-EM, con il segmento DTS che va da una chinasi al sito attivo dell'altra. Quindi hanno eseguito una seconda serie di simulazioni con il segmento DTS rimosso.
"Quello che abbiamo scoperto è che nel sistema senza il segmento distale della coda, l'intero complesso non è stabile, "Spiegava Karandur. "I domini chinasi si muovono rispetto all'impalcatura, il 14-3-3 dimero. In una delle nostre simulazioni, lo stato più debole di B-Raf stesso, che gli esperimenti hanno dimostrato è necessario per mantenere lo stato attivo di questa chinasi, è crollato, indicando che questo segmento distale della coda, DTS, è necessario mantenere effettivamente questo complesso in questa conformazione asimmetrica, che a sua volta è necessario per mantenere il dimero della chinasi nello stato attivo stabile del dimero asimmetrico."
Uno dei principali risultati dello studio è stato trovare il meccanismo d'azione che attiva il complesso chinasi B-Raf di due chinasi B-Raf e due proteine di scaffolding 14-3-3, dove su B-Raf chinasi è l'attivatore, e l'altro è il ricevitore.
"La coda della molecola ricevente è all'interno del sito attivo dell'attivatore, quindi l'attivatore non può funzionare come un enzima, " disse Kondo. "Invece, la molecola attivatore stabilizza la conformazione attiva della molecola ricevente. La proteina dello scaffold 14-3-3 facilita questa disposizione, in modo che l'inserimento della coda avvenga solo per una molecola di chinasi. Ipotizziamo che quando non c'è legame 14-3-3, entrambe le chinasi possono essere bloccate dall'inserimento del DTS, ma questo deve essere testato".
Le sfide computazionali dello studio hanno coinvolto simulazioni di dinamica molecolare che hanno modellato la proteina a livello atomico, determinare le forze di ogni atomo su ogni altro atomo per un sistema di circa 200, 000 atomi a passi temporali di due femtosecondi.
"Per i piccoli impianti, possiamo vedere cosa sta succedendo in tempi relativamente brevi, ma per grandi sistemi come questi, sistemi biomolecolari particolarmente grandi, questi cambiamenti avvengono su scale temporali come i nanosecondi, tempi di microsecondi, o anche tempi di millisecondi, " ha detto Karandur.
Karandur e colleghi si sono rivolti a XSEDE, l'Extreme Science and Engineering Discovery Environment, finanziato dalla NSF, per il tempo di allocazione sul supercomputer Stampede2 presso il Texas Advanced Computing Center (TACC) per eseguire le simulazioni, così come il sistema Bridges al Pittsburgh Supercomputer Center per studiare altre proteine nel percorso. Nodi del processore Skylake di Stampede2, in rete con Intel Omnipath, ha reso rapido il lavoro delle simulazioni di dinamica molecolare NAMD ottimizzate per i supercomputer.
"Stampede2 funziona molto, molto veloce, ed è molto efficiente. Abbiamo generato un totale di circa 1,5 microsecondi di traiettorie per i nostri sistemi in circa quattro-sei settimane. Invece, se l'avessimo eseguito sul nostro cluster interno ci sarebbero voluti mesi o più, " ha detto Karandur.
A proposito di XSEDE, Karandur ha commentato:"Penso che sia una risorsa straordinaria. Ho eseguito simulazioni a partire da quando ero uno studente laureato. XSEDE ci ha permesso di accedere a scale temporali biologicamente rilevanti. Tutto ciò che accade in una cellula, avviene su scale temporali di microsecondi, a tempi di millisecondi, a più a lungo. Quando stavo iniziando, non abbiamo potuto eseguire questa simulazione su nessun sistema da nessuna parte. Voglio dire, ci sarebbero voluti cinque anni, o più. Per poterlo fare in settimane e dire, va bene, sappiamo capire perché questo è importante, quindi ora possiamo iniziare a comprendere meglio come avviene la biologia, è semplicemente fantastico, " ha detto Karandur.
E c'è ancora molto da scoprire su B-Raf. È solo un anello nella catena del segnale che governa la crescita cellulare e il cancro.
"La struttura che è stata risolta in questo documento è parte di un grande, sistema multidominio, " Karandur ha spiegato. "Non sappiamo che aspetto abbia questa proteina completa. Non lo vediamo nella struttura. Non sappiamo come siano le sue dinamiche, e come tutte queste altre parti della proteina svolgono un ruolo nel mantenimento dello stato attivo, o convertendolo dallo stato inattivo allo stato attivo."
Ha sostenuto che man mano che il sistema diventa più grande, i cambiamenti strutturali pertinenti avvengono in tempi più lunghi, e sono necessari supercomputer più grandi per gestire la complessità, come il supercomputer Frontera finanziato dalla NSF, anche al TACC.
"Frontera ci sta arrivando. Siamo molto entusiasti di questo. Siamo in procinto di ottenere un'assegnazione su Frontera, " ha detto Karandur.
Per i non scienziati, questa ricerca fondamentale potrebbe fornire informazioni che porteranno a farmaci migliori per il cancro della pelle.
"L'attivazione paradossale della chinasi Raf da parte di questi inibitori specifici di B-Raf trasforma le cellule normali in tumori durante il trattamento del cancro della pelle, " ha detto Kondo. Comprendere il meccanismo di questo fenomeno ci consentirà di progettare farmaci migliori. il nostro studio può contribuire alla comprensione di questo passaggio. Inoltre, abbiamo trovato mutazioni in questo legame tra il dominio della chinasi e l'elemento di legame 14-3-3 della molecola B-Raf, che non era mai stato mostrato prima. Questa mutazione riduce l'attività di B-Raf nelle cellule. Sta anche indicando che questa parte del dominio della chinasi può essere un punto di riferimento per sviluppare nuovi tipi di inibitori B-Raf".
Karandur ha detto:"Ci sono molte dinamiche che accadono nella cellula. Noi siamo, in gran parte a causa di XSEDE, solo iniziando a essere in grado di guardare cose del genere. Andando avanti, l'unico modo in cui possiamo continuare a guardare le cose è usare molto, supercomputer molto grandi, perché i calcoli richiedono molta potenza di calcolo. È davvero emozionante essere in grado di vedere effettivamente queste cose accadere e dire, ecco come cambiano le cose a livello atomico; ecco queste interazioni tra questi due atomi si formano o si rompono, e questo si traduce in questo enorme cambiamento a livello globale nella struttura complessiva della proteina, e come interagisce con altre proteine, o altre molecole nella cellula. Siamo molto entusiasti di dove andrà in futuro".
Lo studio, "La struttura Cryo-EM di un complesso dimerico B-Raf:14-3-3 rivela l'asimmetria nei siti attivi delle chinasi B-Raf, " è stato pubblicato il 4 ottobre 2019 sulla rivista Scienza .