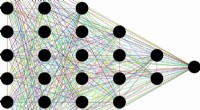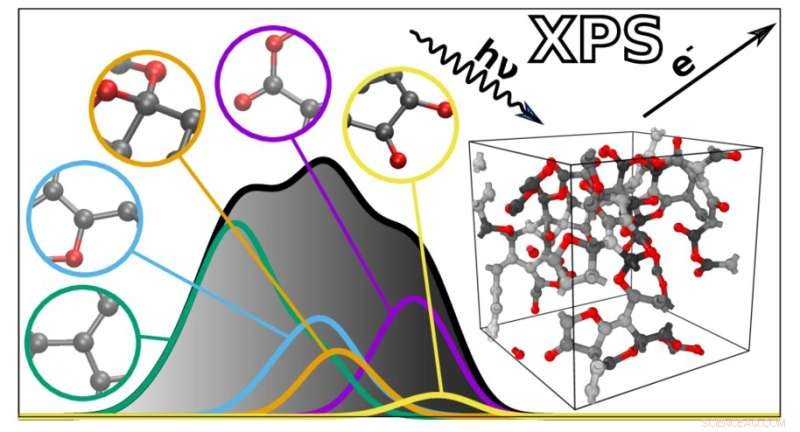
Il nuovo algoritmo prevede gli spettri XPS di materiali complessi in base ai contributi atomici individuali. Credito:Miguel Caro / Università di Aalto
I materiali a base di carbonio hanno un enorme potenziale per la costruzione di un futuro sostenibile, ma gli scienziati dei materiali hanno bisogno di strumenti per analizzare correttamente la loro struttura atomica, che determina le loro proprietà funzionali. La spettroscopia fotoelettronica a raggi X (XPS) è uno degli strumenti utilizzati per farlo, ma i risultati dell'XPS possono essere difficili da interpretare. Ora, i ricercatori di Aalto hanno sviluppato uno strumento di apprendimento automatico per migliorare le analisi XPS, che hanno reso disponibile gratuitamente come XPS Prediction Server.
Gli spettri XPS sono grafici con una raccolta di picchi che riflettono l'energia di legame degli elettroni in profondità negli atomi che compongono un materiale. Poiché le energie di legame dipendono dall'ambiente atomico, possono essere utilizzate per dedurre come gli atomi sono collegati in un particolare materiale o molecola. Tuttavia, questo rende anche gli spettri XPS difficili da interpretare, poiché molti fattori influenzano le energie di legame. Le energie di legame di diverse caratteristiche atomiche possono anche sovrapporsi, complicando ulteriormente l'analisi.
Per aiutare in questo, un team guidato da Miguel Caro ha sviluppato un metodo computazionale in grado di prevedere lo spettro di energia di legame di un materiale basato su un modello strutturale generato dal computer. Ciò semplifica l'interpretazione dei dati XPS consentendo di confrontare le energie di legame osservate sperimentalmente con le previsioni computazionali.
L'idea in sé non è nuova, ma il problema è stato la difficoltà computazionale di calcolare accuratamente lo spettro XPS di un materiale. Il team di Caro ha risolto questo problema utilizzando l'apprendimento automatico. Il trucco consisteva nell'addestrare un algoritmo informatico poco costoso per prevedere il risultato di un metodo di riferimento computazionalmente costoso basato su un'efficiente combinazione di dati meccanici quantistici computazionalmente economici e costosi.
Il metodo computazionalmente più economico, DFT, non corrisponde ai risultati sperimentali in modo molto accurato. Il metodo più accurato, GW, impiega troppo tempo per essere calcolato quando una molecola ha molti atomi. "Abbiamo deciso di costruire un modello di base che utilizza abbondanti dati DFT e quindi perfezionarlo con dati GW rari e preziosi. E ha funzionato", afferma Caro.
L'algoritmo risultante può prevedere lo spettro di qualsiasi materiale disordinato costituito da carbonio, idrogeno e ossigeno. "Gli spettri previsti sono notevolmente vicini a quelli ottenuti sperimentalmente. Ciò apre le porte a una migliore integrazione tra la caratterizzazione sperimentale e computazionale dei materiali", afferma Caro. Successivamente, il team prevede di estendere la propria tecnica per includere una gamma più ampia di materiali e altri tipi di spettroscopia.
L'articolo ad accesso aperto è stato pubblicato in Chimica dei materiali . + Esplora ulteriormente