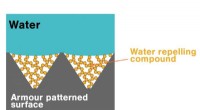
Astratto grafico. Credito:Journal of Chemical Information and Modeling (2022). DOI:10.1021/acs.jcim.2c00389
La simmetria è una caratteristica prevalente della natura a tutte le scale. Ad esempio, i nostri occhi nudi possono facilmente identificare le simmetrie nella forma corporea di innumerevoli organismi. La simmetria è molto importante anche nei campi della fisica e della chimica, specialmente nel regno microscopico degli atomi e delle molecole. I cristalli, che sono materiali altamente ordinati, possono anche avere più tipi di simmetria contemporaneamente, come simmetria rotazionale, simmetria di inversione e simmetria traslazionale.
Ultimamente, insieme ai rapidi progressi dell'informatica, i ricercatori hanno sviluppato metodi computazionali che cercano di prevedere le proprietà fisiche dei cristalli in base alla loro struttura elettronica. In pratica, tuttavia, vengono usati raramente cristalli puri e perfettamente simmetrici. Questo perché le proprietà di un cristallo possono essere regolate a piacimento legandoli con altri materiali o sostituendo casualmente determinati atomi con altri elementi, ad es. drogando.
Di conseguenza, gli scienziati dei materiali stanno cercando approcci computazionalmente efficienti per analizzare tali leghe e cristalli sostituiti, noti anche come soluzioni solide. Il "metodo delle supercelle" è uno di questi approcci ed è ampiamente utilizzato per modellare strutture cristalline con sostituzioni casuali di atomi diversi. La simmetria dei cristalli, tuttavia, è in realtà un problema quando si utilizza questa tecnica. Nei cristalli, possono esserci molti modelli di sostituzione che sono fisicamente equivalenti ad altre sostituzioni se semplicemente li traduciamo o ruotiamo. Trovare questi modelli di sostituzione simmetrica non è molto significativo, e quindi il loro calcolo quando si utilizza il metodo delle supercelle è una perdita di tempo.
In uno studio recente, un team di ricercatori guidato dal professore assistente Kousuke Nakano del Japan Advanced Institute of Science and Technology (JAIST) ha trovato una soluzione a questo problema. Hanno sviluppato un software open source chiamato "Suite per la generazione di modelli ad alto rendimento con sostituzioni atomiche implementate da Python" o SHRY che può, in termini di simmetria, generare modelli di sostituzione distinti in soluzioni solide e leghe. Questo lavoro, che è stato pubblicato nel Journal of Chemical Information and Modeling , è stato co-autore dello studente di dottorato Genki I. Prayogo, del Dr. Andrea Tirelli, del Professor Ryo Maezono e del Professore Associato Kenta Hongo.
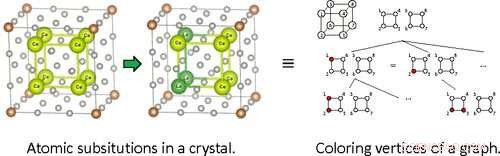
Il team ha affrontato il problema dal punto di vista della teoria dei gruppi. Si scopre che la ricerca di modelli di sostituzione atomica nei cristalli è analoga al problema di trovare modelli di colorazione sui vertici dei grafici sotto determinate restrizioni. Ciò consente di riformulare il problema originale di trovare sostituzioni atomiche non simmetriche nei cristalli esplorando alberi di ricerca che descrivono la colorazione dei vertici nei grafici.
Tuttavia, il modo in cui viene esplorato l'albero di ricerca è cruciale. È impossibile un approccio semplice e ingenuo in cui tutti i possibili rami siano ricercati e confrontati direttamente; il tempo ei calcoli richiesti crescono in modo incontrollabile per i grandi sistemi. Ciò accade perché decidere se esplorare più in basso un ramo richiede informazioni su tutti gli altri rami oltre a quello esplorato, che viene tecnicamente definito "informazioni non locali".
Per evitare questo problema, i ricercatori hanno implementato in SHRY una tecnica chiamata aumento canonico. "Questo metodo può decidere se un ramo di un albero debba essere esplorato in modo più approfondito o meno basandosi esclusivamente su informazioni locali", spiega il dott. Nakano, "Soprattutto, i teoremi della teoria dei gruppi garantiscono che verranno estratti solo modelli di sostituzione distinti, senza sottoesplorare la struttura dell'albero in termini di simmetria." Il team ha verificato che il loro algoritmo fosse privo di errori testandolo a fondo con i dati di un database di strutture cristalline.
Vale la pena notare che SHRY è stato scritto in Python 3, uno dei più popolari linguaggi di programmazione multipiattaforma, e caricato su GitHub, una delle principali piattaforme online per la condivisione di progetti. "SHRY può essere utilizzato come programma autonomo o importato in un altro programma Python come modulo", sottolinea il Dr. Nakano, "Il nostro software utilizza anche il formato CIF (Crystallographic Information File) ampiamente supportato sia per l'input che per l'output del insiemi di strutture cristalline sostituite." Il team prevede di continuare a migliorare il codice di SHRY in base al feedback di altri utenti, aumentandone la velocità e le capacità.
Nel complesso, il software sviluppato in questo studio potrebbe aiutare gli scienziati a identificare potenziali sostituzioni atomiche nei solidi, che è la strategia più comune utilizzata per mettere a punto le proprietà dei materiali per applicazioni pratiche. SHRY aiuterà ad accelerare la ricerca e a sviluppare cristalli sostituiti con funzionalità senza precedenti e caratteristiche superiori. + Esplora ulteriormente