Il team di Jianwei Shuai e quello di Jiahuai Han presso l'Università di Xiamen hanno sviluppato un software di analisi dei dati di acquisizione indipendente dai dati basato su autoencoder profondo per la spettrometria di massa proteica, che realizza l'analisi di peptidi e proteine rilevanti da dati complessi di spettrometria di massa proteica e dimostra la superiorità e versatilità del metodo su diversi strumenti e campioni di specie. Lo studio è stato pubblicato su Research come "Caro-DIA XMBD :codifica automatica profonda per la proteomica di acquisizione indipendente dai dati".
Le proteine svolgono un ruolo fondamentale come esecutori delle attività della vita cellulare, guidando una miriade di processi biologici cruciali. Di conseguenza, il campo della proteomica ha ricevuto ampia attenzione. La proteomica prevede lo studio completo delle proprietà delle proteine, comprese le modifiche post-traduzionali, i livelli di espressione delle proteine, le interazioni proteina-proteina e altro ancora. Il suo obiettivo generale è acquisire una comprensione olistica della patogenesi delle malattie, del metabolismo cellulare e di altri processi vitali a livello proteico.
Tra le tecniche analitiche chiave nella ricerca proteomica, la spettrometria di massa proteica si distingue come la più critica. Nel corso del tempo, la tecnologia della spettrometria di massa si è evoluta per fornire ai ricercatori strumenti affidabili e dinamici per l'analisi proteomica.
Due approcci principali alla spettrometria di massa delle proteine sono l'acquisizione dipendente dai dati (DDA) e l'acquisizione indipendente dai dati (DIA). In DDA, tutti gli spettri di ioni precursori del peptide (MS1) vengono acquisiti in modalità di scansione completa, seguiti dalla selezione degli ioni peptidici con maggiore intensità di N per la frammentazione per ottenere spettri di ioni frammento (MS2).
Nonostante la sua utilità, la DDA deve affrontare sfide legate alla riproducibilità sperimentale e al rilevamento di peptidi a bassa abbondanza a causa della casualità della frammentazione dei peptidi e della selezione preferenziale di peptidi ad alta intensità.
Per superare queste limitazioni è stato introdotto il metodo di acquisizione DIA. Questa tecnica divide l'intervallo del rapporto massa-carica degli spettri degli ioni genitori in più finestre e frammenta sequenzialmente tutti i peptidi all'interno di ciascuna finestra per ottenere spettri degli ioni figli. Un metodo DIA comune è l'acquisizione sequenziale della finestra di tutti gli ioni dei frammenti teorici (SWATH).
Sebbene i dati di acquisizione DIA conservino informazioni proteomiche più complete, le grandi dimensioni dei dati, l'elevata dimensionalità e i segnali spettrali complessi pongono sfide alla sua analisi. Di conseguenza, il data mining DIA è diventato un obiettivo importante nella comunità della proteomica.
Il team di Jianwei Shuai e il team di Jiahuai Han hanno collaborato per sviluppare Dear-DIA, un software di analisi dei dati di acquisizione indipendente dai dati basato sul deep learning, che realizza l'identificazione di frammenti ionici corrispondenti a diversi peptidi da complessi spettri di acquisizione DIA e dimostra la generalizzazione a campioni complessi di specie diverse.
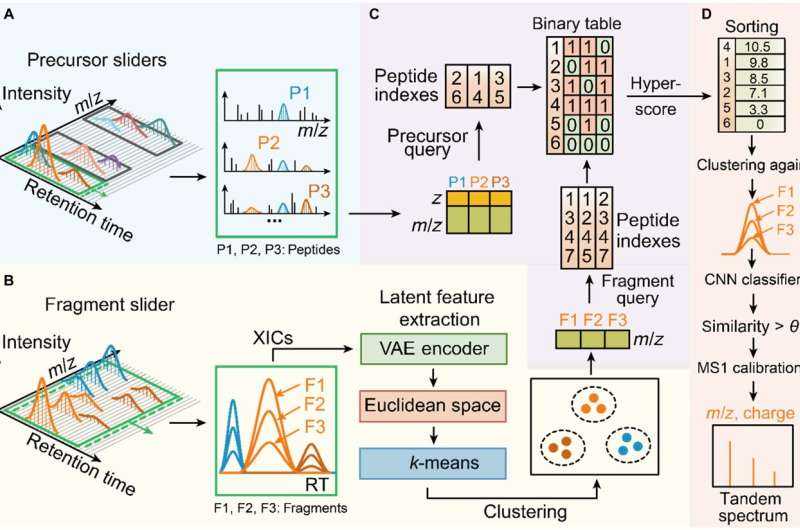
Dear-DIA divide innanzitutto gli spettri in un cursore a larghezza fissa con una larghezza fissa lungo la direzione del tempo di ritenzione (RT) e ciascun cursore contiene un insieme di spettri precursori MS1 e spettri di frammenti MS2 come unità di elaborazione minima. Quindi, è stato utilizzato un algoritmo di ricerca dei picchi per rimuovere gli ioni di fondo con basso rapporto segnale-rumore e trattenere gli ioni precursori candidati e gli ioni frammento candidati.
Successivamente, Dear-DIA utilizza un autocodificatore variazionale per estrarre le caratteristiche di picco degli ioni frammento e mappa le caratteristiche nello spazio euclideo, quindi raggruppa le caratteristiche, con diverse classi di frammenti corrispondenti a diversi peptidi, realizzando così il processo di deconvoluzione dello spettrogramma.
Dear-DIA include un algoritmo di indicizzazione chiamato PIndex, che abbina i precursori ai risultati del clustering dei frammenti e seleziona i migliori risultati di abbinamento in base al punteggio. Dear-DIA utilizza una rete neurale convoluzionale per ricalcolare la somiglianza della forma di picco dei frammenti della stessa classe per eliminare gli ioni interferenti e raggruppare i risultati con bassa somiglianza.
Gli autori hanno prima testato le prestazioni di Dear-DIA su un set di dati SGS Human contenente 422 peptidi sintetici di standard marcati con isotopi stabili divisi in 10 gradienti di diluizione (da 1 a 512 volte di diluizione) e i dati DIA sono stati ottenuti su un AB Spettrometro di massa SCIEX TTOF5600 che utilizza la tecnica SWATH per ottenere dati DIA.
I risultati dell'analisi hanno mostrato che Dear-DIA ha trovato più peptidi sintetici in tutte le soluzioni diluite rispetto ai due metodi analitici comunemente utilizzati, Spectronaut 14 e DIA-Umpire. Gli autori hanno anche confrontato il numero di peptidi e proteine trovati con i diversi metodi analitici per i set di dati SGS Human e L929 Mouse. I risultati hanno mostrato che Dear-DIA è riuscito a trovare più peptidi e proteine rispetto a Spectronaut 14 e DIA-Umpire, coprendo oltre l'85% dei loro risultati.
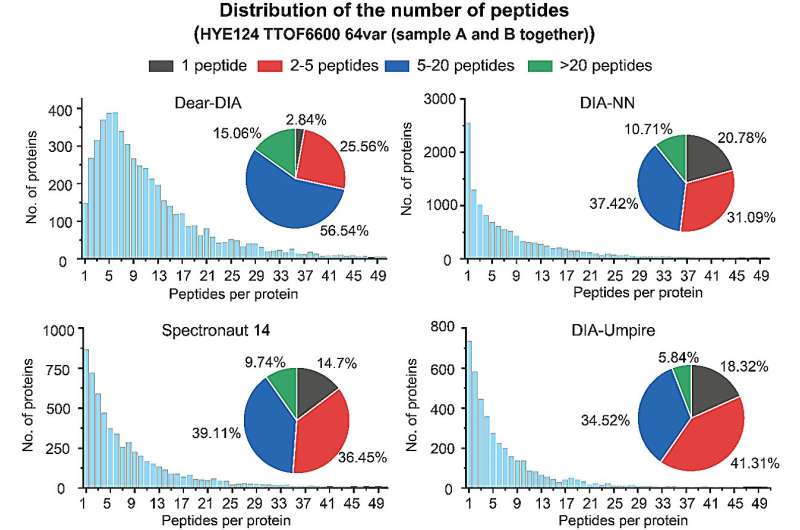
L'affidabilità dei risultati dell'analisi proteomica può essere dimostrata anche dal numero di peptidi identificati per ciascuna proteina. Le proteine con 2 o più peptidi identificati sono generalmente considerate identificazioni più credibili. Gli autori hanno confrontato il numero di proteine rispetto ai peptidi riportati da Dear-DIA con il software esistente su un set di dati di specie miste (set di dati HYE124 TTOF6600 64var).
Il set di dati contiene proteine di tre specie, umana, lievito ed E. coli, e i dati sono stati acquisiti su uno spettrometro di massa AB SCIEX TTOF6600 utilizzando il metodo SWATH, con spettri di ioni principali contenenti 64 finestre variabili. I risultati dell'analisi hanno mostrato che il 97,16% delle proteine trovate da Dear-DIA potrebbero corrispondere a 2 o più peptidi, un valore molto superiore rispetto a DIA-NN, Spectronaut 14 e DIA-Umpire.
Le tecniche di acquisizione indipendente dai dati per la proteomica sono state ampiamente adottate e i relativi algoritmi di analisi sono diventati un punto caldo della ricerca. La scoperta di proteine da dati di spettrometria di massa massiccia è un compito interessante e stimolante. In questo articolo, il team ha sviluppato Dear-DIA, un software di analisi basato sull'apprendimento profondo, che viene utilizzato per elaborare una varietà di dati di acquisizione DIA altamente complessi e può scoprire più peptidi e proteine, oltre a riprodurre la maggior parte dei risultati di Spettronauta e arbitro DIA.
Inoltre, sebbene il set di dati di addestramento provenga da E. coli, le eccellenti prestazioni di Dear-DIA sul set di dati di specie miste dimostrano la sua forte capacità di generalizzazione nell'analisi di dati proteomici complessi. Il deep learning, in quanto strumento ampiamente utilizzato per l'analisi dei big data, ha dimostrato eccellenti capacità di data mining per scoprire profonde associazioni intrinseche nei big data.
L'uso dell'apprendimento profondo per analizzare i dati della spettrometria di massa proteomica ha un grande potenziale e promuoverà ulteriormente lo studio di questioni fondamentali come le reti di segnalazione delle proteine.
Ulteriori informazioni: Qingzu He e altri, caro-DIA XMBD :Deep Autoencoder consente la deconvoluzione della proteomica di acquisizione indipendente dai dati, Ricerca (2023). DOI:10.34133/ricerca.0179
Informazioni sul giornale: Ricerca
Fornito dalla ricerca