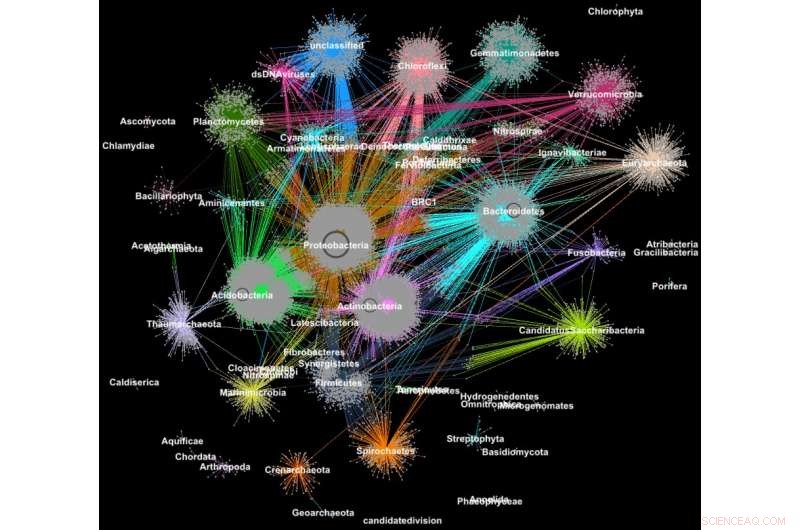
Le proteine dei metagenomi sono raggruppate in famiglie in base alla loro classificazione tassonomica. Credito:Georgios Pavlopoulos e Nikos Kyrpides, JGI/Berkeley Lab
Sapevi che gli strumenti utilizzati per analizzare le relazioni tra gli utenti dei social network o il ranking delle pagine web possono essere estremamente preziosi anche per dare un senso ai big science data? Su un social network come Facebook, ogni utente (persona o organizzazione) è rappresentato come un nodo e le connessioni (relazioni e interazioni) tra di loro sono chiamate bordi. Analizzando queste connessioni, i ricercatori possono imparare molto su ciascun utente:interessi, hobby, abitudini d'acquisto, gli amici, eccetera.
In biologia, algoritmi simili di clustering di grafi possono essere utilizzati per comprendere le proteine che svolgono la maggior parte delle funzioni della vita. Si stima che il solo corpo umano contenga circa 100, 000 diversi tipi di proteine, e quasi tutti i compiti biologici, dalla digestione all'immunità, si verificano quando questi microrganismi interagiscono tra loro. Una migliore comprensione di queste reti potrebbe aiutare i ricercatori a determinare l'efficacia di un farmaco o identificare potenziali trattamenti per una varietà di malattie.
Oggi, tecnologie avanzate ad alto rendimento consentono ai ricercatori di catturare centinaia di milioni di proteine, geni e altri componenti cellulari contemporaneamente e in una serie di condizioni ambientali. Gli algoritmi di clustering vengono quindi applicati a questi set di dati per identificare modelli e relazioni che possono indicare somiglianze strutturali e funzionali. Sebbene queste tecniche siano state ampiamente utilizzate per più di un decennio, non possono tenere il passo con il torrente di dati biologici generati da sequenziatori e microarray di nuova generazione. Infatti, pochissimi algoritmi esistenti possono raggruppare una rete biologica contenente milioni di nodi (proteine) e bordi (connessioni).
Ecco perché un team di ricercatori del Lawrence Berkeley National Laboratory (Berkeley Lab) e del Joint Genome Institute (JGI) del Dipartimento dell'Energia (DOE) ha adottato uno degli approcci di clustering più popolari nella biologia moderna, l'algoritmo Markov Clustering (MCL), e modificato per eseguire rapidamente, efficiente e su larga scala su supercomputer a memoria distribuita. In un caso di prova, il loro algoritmo ad alte prestazioni, chiamato HipMCL, ha raggiunto un'impresa precedentemente impossibile:raggruppare una grande rete biologica contenente circa 70 milioni di nodi e 68 miliardi di bordi in un paio d'ore, utilizzando circa 140, 000 core di processore sul supercomputer Cori del National Energy Research Scientific Computing Center (NERSC). Un articolo che descrive questo lavoro è stato recentemente pubblicato sulla rivista Ricerca sugli acidi nucleici .
"Il vero vantaggio di HipMCL è la sua capacità di raggruppare enormi reti biologiche che erano impossibili da raggruppare con il software MCL esistente, permettendoci così di identificare e caratterizzare il nuovo spazio funzionale presente nelle comunità microbiche, "dice Nikos Kyrpides, che dirige gli sforzi di Microbiome Data Science di JGI e il Prokaryote Super Program ed è coautore del documento. "Inoltre possiamo farlo senza sacrificare la sensibilità o l'accuratezza del metodo originale, che è sempre la sfida più grande in questo tipo di sforzi di ridimensionamento."
"Man mano che i nostri dati crescono, sta diventando ancora più imperativo spostare i nostri strumenti in ambienti di elaborazione ad alte prestazioni, " aggiunge. "Se mi chiedessi quanto è grande lo spazio proteico? La verità è, non lo sappiamo davvero perché fino ad ora non avevamo gli strumenti computazionali per raggruppare efficacemente tutti i nostri dati genomici e sondare la materia oscura funzionale".
Oltre ai progressi nella tecnologia di raccolta dei dati, i ricercatori scelgono sempre più di condividere i propri dati in database comunitari come il sistema Integrated Microbial Genomes &Microbiomes (IMG/M), che è stato sviluppato attraverso una collaborazione decennale tra scienziati del JGI e la divisione di ricerca computazionale (CRD) del Berkeley Lab. Ma consentendo agli utenti di fare analisi comparative ed esplorare le capacità funzionali delle comunità microbiche in base alla loro sequenza metagenomica, Anche gli strumenti della comunità come IMG/M stanno contribuendo all'esplosione dei dati nella tecnologia.
In che modo le passeggiate casuali portano a colli di bottiglia nell'informatica
Per avere un controllo su questo torrente di dati, i ricercatori si affidano all'analisi dei cluster, o raggruppamento. Questo è essenzialmente il compito di raggruppare gli oggetti in modo che gli elementi nello stesso gruppo (cluster) siano più simili di quelli in altri cluster. Da più di un decennio, i biologi computazionali hanno favorito l'MCL per raggruppare le proteine per somiglianze e interazioni.
"Uno dei motivi per cui MCL è stato popolare tra i biologi computazionali è che è relativamente privo di parametri; gli utenti non devono impostare un sacco di parametri per ottenere risultati accurati ed è notevolmente stabile a piccole alterazioni nei dati. Questo è importante perché potrebbe essere necessario ridefinire una somiglianza tra i punti dati o potrebbe essere necessario correggere un leggero errore di misurazione nei dati. In questi casi, non vuoi che le tue modifiche cambino l'analisi da 10 cluster a 1, 000 cluster, "dice Aydin Buluç, uno scienziato CRD e uno dei coautori del documento.
Ma, Aggiunge, la comunità della biologia computazionale sta incontrando un collo di bottiglia informatico perché lo strumento viene eseguito principalmente su un singolo nodo di computer, è computazionalmente costoso da eseguire e ha un grande footprint di memoria, il che limita la quantità di dati che questo algoritmo può raggruppare.
Uno dei passaggi più impegnativi dal punto di vista del calcolo e della memoria in questa analisi è un processo chiamato random walk. Questa tecnica quantifica la forza di una connessione tra nodi, che è utile per classificare e prevedere i collegamenti in una rete. Nel caso di una ricerca su Internet, questo potrebbe aiutarti a trovare una camera d'albergo economica a San Francisco per le vacanze di primavera e anche a dirti il momento migliore per prenotarla. In biologia, un tale strumento potrebbe aiutarti a identificare le proteine che stanno aiutando il tuo corpo a combattere un virus influenzale.
Dato un grafo o una rete arbitraria, è difficile conoscere il modo più efficiente per visitare tutti i nodi ei collegamenti. Una passeggiata casuale ottiene un senso dell'impronta esplorando l'intero grafico in modo casuale; inizia da un nodo e si sposta arbitrariamente lungo un bordo fino a un nodo vicino. Questo processo continua finché non sono stati raggiunti tutti i nodi sulla rete del grafo. Poiché ci sono molti modi diversi di viaggiare tra i nodi di una rete, questo passaggio si ripete numerose volte. Algoritmi come MCL continueranno a eseguire questo processo di passeggiata casuale fino a quando non ci sarà più una differenza significativa tra le iterazioni.
In una data rete, potresti avere un nodo connesso a centinaia di nodi e un altro nodo con una sola connessione. Le passeggiate casuali cattureranno i nodi altamente connessi perché verrà rilevato un percorso diverso ogni volta che viene eseguito il processo. Con queste informazioni, l'algoritmo può prevedere con un livello di certezza come un nodo della rete è connesso ad un altro. Tra ogni corsa casuale, l'algoritmo segna la sua previsione per ogni nodo sul grafico in una colonna di una matrice di Markov, una specie di libro mastro, e alla fine vengono rivelati i cluster finali. Sembra abbastanza semplice, ma per le reti proteiche con milioni di nodi e miliardi di bordi, questo può diventare un problema estremamente computazionale e ad alta intensità di memoria. Con HipMCL, Gli scienziati informatici del Berkeley Lab hanno utilizzato strumenti matematici all'avanguardia per superare queste limitazioni.
"Abbiamo in particolare mantenuto intatto il backbone MCL, rendendo HipMCL un'implementazione massicciamente parallela dell'algoritmo MCL originale, "dice Ariful Azad, un informatico in CRD e autore principale dell'articolo.
Sebbene ci siano stati precedenti tentativi di parallelizzare l'algoritmo MCL per l'esecuzione su una singola GPU, lo strumento potrebbe ancora raggruppare solo reti relativamente piccole a causa delle limitazioni di memoria su una GPU, Appunti di Azad.
"Con HipMCL essenzialmente rielaborare gli algoritmi MCL per funzionare in modo efficiente, in parallelo su migliaia di processori, e configurarlo per sfruttare la memoria aggregata disponibile in tutti i nodi di calcolo, " aggiunge. "La scalabilità senza precedenti di HipMCL deriva dall'uso di algoritmi all'avanguardia per la manipolazione di matrici sparse".
Secondo Buluc, l'esecuzione simultanea di una passeggiata casuale da molti nodi del grafico è calcolata al meglio utilizzando la moltiplicazione di matrici a matrice sparsa, che è una delle operazioni più basilari nello standard GraphBLAS recentemente rilasciato. Buluç e Azad hanno sviluppato alcuni degli algoritmi paralleli più scalabili per la moltiplicazione di matrici a matrice sparsa di GraphBLAS e hanno modificato uno dei loro algoritmi all'avanguardia per HipMCL.
"Il punto cruciale qui era trovare il giusto equilibrio tra parallelismo e consumo di memoria. HipMCL estrae dinamicamente quanto più parallelismo possibile data la memoria disponibile ad esso assegnata, "dice Buluc.
HipMCL:Clustering su larga scala
Oltre alle innovazioni matematiche, un altro vantaggio di HipMCL è la sua capacità di funzionare senza problemi su qualsiasi sistema, inclusi laptop, workstation e grandi supercomputer. I ricercatori hanno raggiunto questo obiettivo sviluppando i propri strumenti in C++ e utilizzando librerie standard MPI e OpenMP.
"Abbiamo ampiamente testato HipMCL su Intel Haswell, Processori Ivy Bridge e Knights Landing al NERSC, utilizzando un massimo di 2, 000 nodi e mezzo milione di thread su tutti i processori, e in tutte queste esecuzioni HipMCL ha raggruppato con successo reti in cluster comprendenti da migliaia a miliardi di bordi, " dice Buluç. "Vediamo che non c'è barriera nel numero di processori che può utilizzare per eseguire e scopriamo che può raggruppare le reti 1, 000 volte più veloce dell'algoritmo MCL originale."
"HipMCL sarà davvero trasformativo per la biologia computazionale dei big data, proprio come i sistemi IMG e IMG/M sono stati per la genomica del microbioma, " dice Kyrpides. "Questo risultato è una testimonianza dei vantaggi della collaborazione interdisciplinare al Berkeley Lab. Come biologi comprendiamo la scienza, ma è stato così prezioso poter collaborare con scienziati informatici che possono aiutarci ad affrontare i nostri limiti e spingerci avanti".
Il loro prossimo passo è continuare a rielaborare HipMCL e altri strumenti di biologia computazionale per futuri sistemi exascale, che sarà in grado di calcolare quintilioni di calcoli al secondo. Ciò sarà essenziale poiché i dati genomici continuano a crescere a un ritmo sbalorditivo, raddoppiando circa ogni cinque o sei mesi. Questo sarà fatto come parte del centro di co-design Exagraph del DOE Exascale Computing Project.