
Shantenu Jha, presidente del Center for Data-Driven Discovery del Brookhaven Lab, e il suo team della Rutgers University e dell'University College di Londra hanno progettato un framework software per calcolare in modo accurato e rapido quanto fortemente i candidati ai farmaci si legano alle loro proteine bersaglio. Il quadro mira a risolvere il problema del mondo reale della progettazione di farmaci, attualmente un processo lungo e costoso, e potrebbe avere un impatto sulla medicina personalizzata. Credito:Brookhaven National Laboratory
Le soluzioni a molti problemi scientifici e ingegneristici del mondo reale, dal miglioramento dei modelli meteorologici alla progettazione di nuovi materiali energetici per comprendere come si è formato l'universo, richiedono applicazioni in grado di raggiungere dimensioni molto grandi e prestazioni elevate. Ogni anno, attraverso la sua International Scalable Computing Challenge (SCALE), l'Institute of Electrical and Electronics Engineers (IEEE) riconosce un progetto che fa avanzare lo sviluppo di applicazioni e l'infrastruttura di supporto per consentire calcolo ad alte prestazioni necessario per risolvere tali problemi.
Il vincitore di quest'anno, "Consentire il compromesso tra accuratezza e costo computazionale:algoritmi adattivi per ridurre i tempi di analisi clinica, " è il risultato di una collaborazione tra chimici e scienziati computazionali e informatici presso il Brookhaven National Laboratory del Dipartimento dell'Energia degli Stati Uniti (DOE), Università di Rutger, e l'University College di Londra. I membri del team sono stati premiati al 18th IEEE/Association for Computing Machinery (ACM) International Symposium on Cluster, Cloud and Grid Computing tenutosi a Washington, DC, dal 1 al 4 maggio
"Abbiamo sviluppato una metodologia di calcolo numerico per valutare in modo accurato e rapido l'efficacia di diversi farmaci candidati, " ha detto il membro del team Shantenu Jha, presidente del Centro per la scoperta guidata dai dati, parte della Computational Science Initiative di Brookhaven Lab. "Anche se non abbiamo ancora applicato questa metodologia per progettare un nuovo farmaco, abbiamo dimostrato che potrebbe funzionare su larga scala coinvolta nel processo di scoperta dei farmaci".
La scoperta di farmaci è un po' come progettare una chiave per inserire un lucchetto. Affinché un farmaco sia efficace nel trattamento di una particolare malattia, deve legarsi strettamente a una molecola, di solito una proteina, che è associata a quella malattia. Solo allora il farmaco può attivare o inibire la funzione della molecola bersaglio. I ricercatori possono selezionare 10, 000 o più composti molecolari prima di trovarne uno che abbia l'attività biologica desiderata. Ma questi composti "di piombo" spesso mancano della potenza, selettività, o stabilità necessaria per diventare una droga. Modificando la struttura chimica di questi conduttori, i ricercatori possono progettare composti con le appropriate proprietà simili a farmaci. I candidati ai farmaci progettati si spostano quindi lungo la pipeline di sviluppo fino alla fase di test preclinici. Di questi candidati, solo una piccola parte entra nella fase di sperimentazione clinica, e solo uno finisce per diventare un farmaco approvato per l'uso del paziente. Portare un nuovo farmaco sul mercato può richiedere un decennio o più e costare miliardi di dollari.
Superare i colli di bottiglia nella progettazione dei farmaci attraverso la scienza computazionale
I recenti progressi nella tecnologia e nella conoscenza hanno portato a una nuova era di scoperta di farmaci, che potrebbe ridurre significativamente i tempi e le spese del processo di sviluppo dei farmaci. I miglioramenti nella nostra comprensione delle strutture cristalline 3-D delle molecole biologiche e l'aumento della potenza di calcolo stanno rendendo possibile l'uso di metodi computazionali per prevedere le interazioni farmaco-bersaglio.
La scoperta di farmaci è un problema di blocco e chiave in cui il farmaco (chiave) deve adattarsi specificamente al bersaglio biologico (blocco). Credito:Brookhaven National Laboratory
In particolare, una tecnica di simulazione al computer chiamata dinamica molecolare si è dimostrata promettente nel prevedere con precisione la forza con cui le molecole dei farmaci si legano ai loro bersagli (affinità di legame). La dinamica molecolare simula il modo in cui gli atomi e le molecole si muovono mentre interagiscono nel loro ambiente. In caso di scoperta di farmaci, le simulazioni rivelano come le molecole dei farmaci interagiscono con la loro proteina bersaglio e cambiano la conformazione della proteina, o forma, che ne determina la funzione.
Però, queste capacità di previsione non operano ancora su una scala sufficientemente ampia o a una velocità sufficientemente rapida da consentire alle aziende farmaceutiche di adottarle nel loro processo di sviluppo.
"Tradurre questi progressi nell'accuratezza predittiva per avere un impatto sul processo decisionale industriale richiede che nell'ordine di 10, 000 affinità di legame vengono calcolate il più rapidamente possibile, senza perdita di precisione, " ha affermato Jha. "La produzione di informazioni tempestive richiede un'efficienza computazionale basata sullo sviluppo di nuovi algoritmi e sistemi software scalabili, e l'allocazione intelligente delle risorse di supercalcolo."
Jha e i suoi collaboratori alla Rutgers University, dove è anche docente presso il Dipartimento di Ingegneria Elettrica e Informatica, e l'University College di Londra hanno progettato un framework software per supportare il calcolo accurato e rapido delle affinità di legame ottimizzando l'uso delle risorse computazionali. Questo quadro, chiamato High-Throughput Binding Affinity Calculator (HTBAC), si basa sul progetto RADICAL-Cybertools che Jha guida come ricercatore principale della ricerca di Rutgers in Advanced Distributed Cyberinfrastructure and Applications Laboratory (RADICAL). L'obiettivo di RADICAL-Cybertools è fornire una suite di componenti software per supportare i flussi di lavoro di applicazioni scientifiche su larga scala su piattaforme di calcolo ad alte prestazioni, che aggregano la potenza di calcolo per risolvere grandi problemi computazionali che altrimenti sarebbero irrisolvibili a causa del tempo necessario.
Nell'informatica, i flussi di lavoro si riferiscono a una serie di passaggi di elaborazione necessari per completare un'attività o risolvere un problema. Soprattutto per i flussi di lavoro scientifici, è importante che i flussi di lavoro siano flessibili in modo che possano adattarsi dinamicamente durante il runtime per fornire i risultati più accurati facendo un uso efficiente del tempo di calcolo disponibile. Tali flussi di lavoro adattivi sono ideali per la scoperta di farmaci perché solo i farmaci con elevate affinità di legame dovrebbero essere ulteriormente valutati.
"Il compromesso desiderato tra l'accuratezza richiesta e il costo computazionale (tempo) cambia durante la scoperta del farmaco mentre il processo passa dallo screening alla selezione dei lead e quindi all'ottimizzazione dei lead, " ha affermato Jha. "Un numero significativo di composti deve essere sottoposto a screening a basso costo per eliminare i leganti scadenti prima che siano necessari metodi più accurati per discriminare i leganti migliori. Fornire il più rapido tempo possibile per la soluzione richiede il monitoraggio dei progressi delle simulazioni e la base delle decisioni sulla continuazione dell'esecuzione sulla rilevanza scientifica".
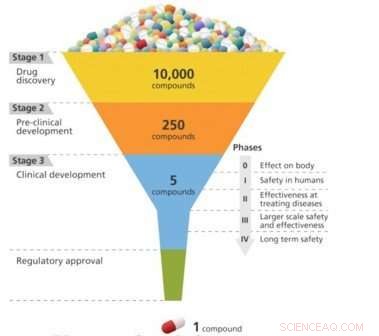
Uno schema del processo di sviluppo del farmaco, che progressivamente affina i candidati più efficaci da un ampio pool iniziale. Credito:Brookhaven National Laboratory
In altre parole, non avrebbe senso continuare le simulazioni di una particolare interazione farmaco-proteina se il farmaco si lega debolmente alla proteina rispetto agli altri candidati. Ma avrebbe senso allocare risorse computazionali aggiuntive se un farmaco mostra un'elevata affinità di legame.
Il supporto di flussi di lavoro adattivi su larga scala caratteristici dei programmi di scoperta di farmaci richiede capacità computazionali avanzate. HTBAC fornisce tale supporto attraverso un livello software middleware flessibile che consente l'esecuzione adattiva di algoritmi. Attualmente, HTBAC supporta due algoritmi:campionamento avanzato della dinamica molecolare con approssimazione del solvente continuo (ESMACS) e integrazione termodinamica con campionamento avanzato (TIES). ESMACS, un metodo computazionalmente più economico ma meno rigoroso di TIES, calcola la forza di legame di un farmaco alla sua proteina bersaglio sulla base di simulazioni di dinamica molecolare. Al contrario, TIES confronta le affinità di legame relative di due farmaci diversi con la stessa proteina.
"ESMACS fornisce un approccio quantitativo rapido abbastanza sensibile da determinare le affinità di legame in modo da poter eliminare i leganti poveri, mentre TIES fornisce un metodo più accurato per studiare i buoni leganti man mano che vengono perfezionati e migliorati, " disse Jumana Dakka, un dottorato di ricerca del secondo anno studente alla Rutgers e membro del gruppo RADICAL.
Per determinare quale algoritmo eseguire, HTBAC analizza i calcoli dell'affinità di legame in fase di esecuzione. Questa analisi informa le decisioni sul numero di simulazioni simultanee da eseguire e se i passaggi di stimolazione devono essere aggiunti o rimossi per ciascun candidato farmaco indagato.
Mettere alla prova il quadro
Jha's team demonstrated how HTBAC could provide insight from drug candidate data on a short timescale by reproducing results from a collaborative study between University College London and the London-based pharmaceutical company GlaxoSmithKline to discover drug compounds that bind to the BRD4 protein. Known to play a key role in driving cancer and inflammatory diseases, the BRD4 protein is a major target of bromodomain-containing (BRD) inhibitors, a class of pharmaceutical drugs currently being evaluated in clinical trials. The researchers involved in this collaborative study are focusing on identifying promising new drugs to treat breast cancer while developing an understanding of why certain drugs fail in the presence of breast cancer gene mutations.
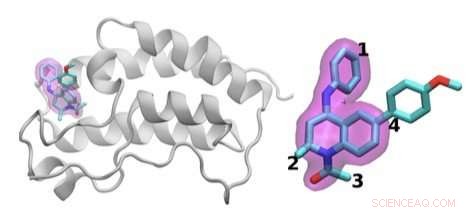
The scientists investigated the chemical structures of 16 drugs based on the same tetrahydroquinoline (THQ) scaffold. On the left is a cartoon of the BRD4 protein bound to one of these drugs; on the right is a molecular representation of a drug with the THQ scaffold highlighted in magenta. Regions that are chemically modified between the drugs investigated in this study are labeled 1 to 4. Typically, only a small change is made to the chemical structure of one drug to the next. This conservative approach makes it easier for researchers to understand why one drug is effective, whereas another is not. Credito:Brookhaven National Laboratory
Jha and his team concurrently screened a group of 16 closely related drug candidates from the study by running thousands of computational sequences on more than 32, 000 computing cores. They ran the computations on the Blue Waters supercomputer at the National Center for Computing Applications, University of Illinois at Urbana-Champaign.
In a real drug design scenario, many more compounds with a wider range of chemical properties would need to be investigated. The team members previously demonstrated that the workload management layer and runtime system underlying HTBAC could scale to handle 10, 000 concurrent tasks.
"HTBAC could support the concurrent screening of different compounds at unprecedented scales—both in the number of compounds and computational resources used, " said Jha. "We showed that HTBAC has the ability to solve a large number of drug candidates in essentially the same amount of time it would take to solve a smaller set, assuming the number of processors increases proportionally with the number of candidates."
This ability is made possible through HTBAC's adaptive functionality, which allows it to execute the optimal algorithm depending on the properties of the drugs being investigated, improving the accuracy of the results and minimizing compute time.
"The lead optimization stage usually considers on the order of 10, 000 small molecules, " said Jha. "While experiment automation reduces the amount of time needed to calculate the binding affinities, HTBAC has the potential to cut this time (and cost) by an order of magnitude or more."
With HTBAC, TIES requires approximately 25, 000 central processing unit (CPU) core hours for a single prediction. At least a 250 million core hours would be needed for a large-scale study to support a pharmaceutical drug screening campaign, with a typical turnaround time of about two weeks. HTBAC could facilitate running studies requiring sustained usage of millions of core hours per day.
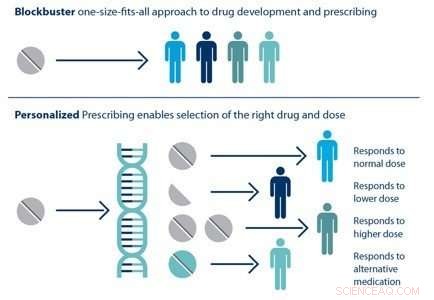
Individual patients respond differently to drugs. The ability to predict which treatment is best for a particular patient based on his or her genetic sequence is the goal of personalized medicine. Credito:Brookhaven National Laboratory
When the University of College London–GlaxoSmithKline study concludes, Jha and his team hope to be given the experimental data on the tens of thousands of drug candidates, without knowing which candidate ended up being the best one. Con queste informazioni, they could perform a blind test to determine whether HTBAC provides an improvement in compute time (for a given accuracy) over the existing automated methods for drug discovery. Se necessario, they could then refine their methodology.
Applying scalable computing to precision medicine
HTBAC not only has the potential to improve the speed and accuracy of drug discovery in the pharmaceutical industry but also to improve individual patient outcomes in clinical settings. Using target proteins based on a patient's genetic sequence, HTBAC could predict a patient's response to different drug treatments. This personalized assessment could replace the traditional one-size-fits-all approach to medicine. Per esempio, such predictions could help determine which cancer patients would actually benefit from chemotherapy, avoiding unnecessary toxicity.
According to Jha, the computation time would have to be significantly reduced in order for physicians to clinically use HTBAC to treat their patients:"Our grand vision is to apply scalable computing techniques to personalized medicine. If we can use these techniques to optimize drugs and drug cocktails for each individual's unique genetic makeup on the order of a few days, we will be empowered to treat diseases much more effectively."
"Extreme-scale computing for precision medicine is an emerging area that CSI and Brookhaven at large have begun to tackle, " said CSI Director Kerstin Kleese van Dam. "This work is a great example of how technologies we originally developed to tackle DOE challenges can be applied to other domains of high national impact. We look forward to forming more strategic partnerships with other universities, pharmaceutical companies, and medical institutions in this important area that will transform the future of health care."














