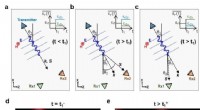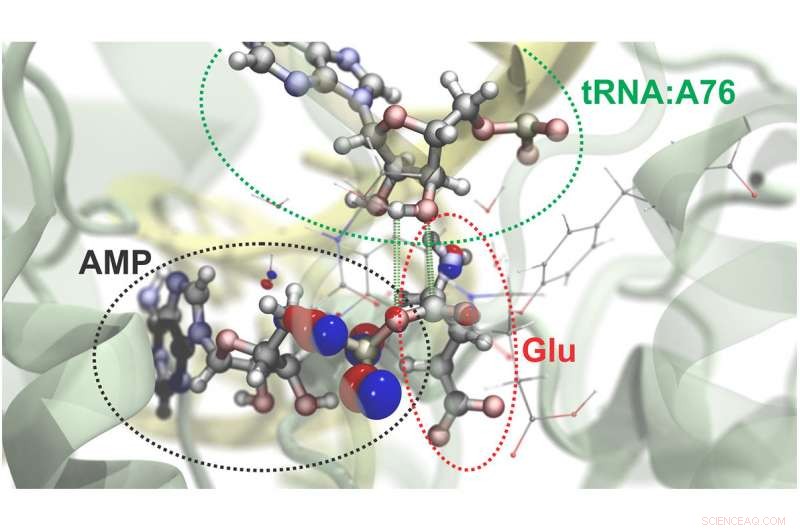
I ricercatori possono simulare le dinamiche atomiche e subatomiche in grandi sistemi molecolari. Ecco una visualizzazione del processo mediante il quale l'amminoacido glutammato (Glu) è attaccato a una regione specifica del suo RNA di trasferimento (tRNA). Una molecola ricca di energia, ATP, guida questa reazione e viene convertito in AMP nel processo. Le bolle rosse e blu rappresentano la probabilità di trovare elettroni in particolari regioni. Le linee tratteggiate verdi delineano gli atomi che si legano in questa reazione chimica. Credito:Rafael Bernardi, Zan Luthey-Schulten e Marcelo Melo
Gli scienziati hanno costruito un "microscopio computazionale" in grado di simulare le forze atomiche e subatomiche che guidano le interazioni molecolari. Questo strumento semplificherà gli sforzi per comprendere la chimica della vita, modellare grandi sistemi molecolari e sviluppare nuovi agenti farmaceutici e industriali, dicono i ricercatori.
Riportano i loro risultati sulla rivista Metodi della natura .
Gli scienziati hanno combinato due approcci computazionali utilizzati per simulare le interazioni molecolari. Il primo, un programma di dinamica molecolare su nanoscala noto come NAMD, utilizza metodi della meccanica classica per modellare la struttura e simulare il comportamento di centinaia di milioni di singoli atomi. Il secondo programma ingrandisce il regno subatomico, simulando le interazioni dei protoni, neutroni ed elettroni. La modellazione su questa scala quantomeccanica richiede molta potenza di calcolo, così i ricercatori hanno implementato un metodo per suddividere grandi molecole in regioni di meccanica classica e quantistica. Ciò consente loro di concentrare le proprie risorse computazionali su piccole regioni coinvolte in interazioni critiche, come la creazione o la rottura di legami chimici.
Sia i programmi di meccanica molecolare che quelli di meccanica quantistica sono disponibili da anni, e altri team hanno lavorato per combinarli, ha detto il professore di chimica dell'Università dell'Illinois Zaida (Zan) Luthey-Schulten, che ha condotto la nuova ricerca con il marito, Università di I. professore di fisica Klaus Schulten. Ma il nuovo sforzo semplifica il processo di creazione, eseguire e analizzare le simulazioni.
"L'abbiamo impostato in modo che i ricercatori possano facilmente scegliere come partizionare i propri sistemi, "Luthey-Schulten ha detto. "I miei studenti lo stanno provando, e la maggior parte di loro è in grado di farlo senza troppe difficoltà."
Schulten ha sviluppato NAMD in Illinois nel 1995, combinandolo con un software di visualizzazione, VMD, che consente ai ricercatori di osservare lo svolgersi delle interazioni molecolari su larga scala. Schulten, morto nel 2016 ha equiparato questo approccio alla "costruzione di un microscopio computazionale".
Il microscopio computazionale è ideale per modellare tratti strutturali e movimenti di grandi complessi. Per esempio, nel 2013, Schulten e i suoi colleghi hanno usato la NAMD per modellare il capside dell'HIV, che è composto da più di 1, 300 proteine identiche che si assemblano in una struttura a gabbia che protegge il virus finché non entra in una cellula ospite. Quella simulazione ha rappresentato le interazioni di oltre 64 milioni di atomi e ha richiesto l'uso del supercomputer Blue Waters presso il National Center for Supercomputing Applications presso l'U. of I. Il nuovo studio ha utilizzato anche Blue Waters, questa volta per migliorare la risoluzione del microscopio computazionale.

Da sinistra, dottorando Marcelo Melo, professoressa di chimica Zaida Luthey-Schulten, il ricercatore post-dottorato Rafael Bernardi ei suoi colleghi hanno sviluppato un nuovo approccio alla modellazione di grandi interazioni molecolari su scala atomica e subatomica. Il loro lavoro semplifica il metodo per altri scienziati e studenti. Credito:L. Brian Stauffer
Il software NAMD è progettato per descrivere il comportamento dei singoli atomi. Ma i singoli atomi coinvolti in interazioni e reazioni chimiche specifiche non si comportano sempre come le loro controparti altrove. Capire come variano richiede uno sguardo più da vicino alle forze subatomiche in gioco. Ciò è particolarmente importante nelle regioni dinamiche delle molecole, ad esempio quei luoghi dove si creano o si rompono i legami chimici, hanno detto i ricercatori.
Nel nuovo studio, il team di ricerca dell'Illinois ha collaborato con gli esperti di QM Frank Neese, dell'Istituto Max Planck per la ricerca sul carbone di Mulheim an der Ruhr, Germania; e Gerd B. Rocha, dell'Università Federale di Paraiba, a João Pessoa, Brasile.
A dimostrazione del nuovo approccio, i ricercatori hanno simulato il comportamento chimico degli RNA di trasferimento, molecole che svolgono un ruolo chiave nella traduzione delle informazioni genetiche in proteine. Usando NAMD, hanno modellato la struttura molecolare complessiva del tRNA nel momento in cui una proteina speciale carica un amminoacido sul tRNA. Hanno suddiviso due siti del complesso in regioni che richiedono un approccio quantomeccanico più mirato. (Guarda un film della simulazione.)
Le simulazioni subatomiche delle interazioni delle due regioni hanno permesso al team di eseguire simulazioni di quattro diversi scenari che consentirebbero al tRNA di funzionare come fa nella cellula. Le loro simulazioni hanno rivelato che uno dei quattro potenziali percorsi chimici era energeticamente più favorevole degli altri e quindi più probabile che si verificasse.
I ricercatori hanno anche utilizzato vari metodi per partizionare il complesso del tRNA tra le regioni MM e QM e hanno riferito su ciascun approccio.
"Non abbiamo scelto un solo modo; ne abbiamo scelti il maggior numero possibile. Diamo libertà all'utente. Il modo in cui lo strutturi dipende davvero dal particolare sistema che stai studiando, " ha detto il ricercatore postdottorato dell'Università di I. Rafael Bernardi, un co-autore principale dello studio con lo studente laureato Marcelo Melo.
"Non facciamo l'intero sistema in modo quantistico perché ci vorrebbe un'eternità per calcolare, " disse Melo.
"NAMD è stato progettato - e questa era la visione di mio marito - per trattare sistemi davvero grandi, "Luthey-Schulten ha detto. "Ora possiamo aggiungere la scala subatomica a quella, aprendo nuove e vaste possibilità di ricerca”.