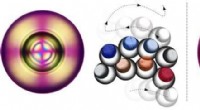
Rappresentazione artistica della struttura atomica del cristallo di carburo di silicio che mostra il difetto (cerchio viola) e la regione di interesse identificata con la teoria della meccanica quantistica (sfera d'argento). Credito:Università di Chicago
I computer quantistici hanno un enorme potenziale per i calcoli che utilizzano nuovi algoritmi e coinvolgono quantità di dati ben oltre la capacità dei supercomputer di oggi. Mentre tali computer sono stati costruiti, sono ancora agli inizi e hanno un'applicabilità limitata per risolvere problemi complessi nella scienza dei materiali e nella chimica. Per esempio, consentono solo la simulazione delle proprietà di pochi atomi per la ricerca sui materiali.
Gli scienziati dell'Argonne National Laboratory del Dipartimento dell'Energia degli Stati Uniti (DOE) e dell'Università di Chicago (UCicago) hanno sviluppato un metodo che apre la strada all'utilizzo di computer quantistici per simulare molecole realistiche e materiali complessi, la cui descrizione richiede centinaia di atomi.
Il gruppo di ricerca è guidato da Giulia Galli, direttore del Midwest Integrated Center for Computational Materials (MICCoM), un leader di gruppo nella divisione Scienza dei materiali di Argonne e membro del Centro per l'ingegneria molecolare ad Argonne. Galli è anche professore di struttura elettronica e simulazioni della famiglia Liew presso la Pritzker School of Molecular Engineering e professore di chimica all'UChicago. Ha lavorato a questo progetto con l'assistente scienziato Marco Govoni e lo studente laureato He Ma, entrambi parte della divisione Materials Science di Argonne e UChicago.
"Il nostro metodo di calcolo di nuova concezione, "Galli ha detto, "migliora notevolmente l'accuratezza ottenibile con i metodi quantomeccanici esistenti per quanto riguarda i calcoli per difetti specifici nei materiali cristallini, e l'abbiamo implementato su un computer quantistico."
Negli ultimi tre decenni, gli approcci teorici della meccanica quantistica hanno svolto un ruolo importante nella previsione delle proprietà dei materiali rilevanti per la scienza dell'informazione quantistica e dei materiali funzionali per le applicazioni energetiche, che comprende catalizzatori e sistemi di accumulo di energia. Però, questi approcci sono computazionalmente impegnativi, ed è ancora difficile applicarli a complessi, materiali eterogenei.
"Nella nostra ricerca abbiamo sviluppato una teoria dell'incorporamento quantistico che ha permesso la simulazione di "difetti di spin" nei solidi accoppiando hardware di calcolo quantistico e classico, " Ha detto Govoni. Questi tipi di difetti nei solidi hanno applicabilità allo sviluppo di materiali per l'elaborazione delle informazioni quantistiche e applicazioni di rilevamento su scala nanometrica ben oltre le attuali capacità.
"La nostra è una potente strategia lungimirante nella scienza computazionale dei materiali con il potenziale di prevedere le proprietà di materiali complessi in modo più accurato di quanto i metodi attuali più avanzati possano fare al momento, " ha aggiunto Govoni.
Il team ha prima testato il metodo di inclusione quantistica su un computer classico, applicandolo ai calcoli delle proprietà dei difetti di spin nel diamante e nel carburo di silicio. "I ricercatori del passato hanno studiato a fondo i difetti sia nel diamante che nel carburo di silicio, quindi avevamo abbondanti dati sperimentali da confrontare con le previsioni del nostro metodo, " ha detto Ma. Il buon accordo tra teoria ed esperimento ha dato al team fiducia nell'affidabilità del loro metodo.
Il team è poi passato a testare gli stessi calcoli su un simulatore quantistico e infine sul computer quantistico IBM Q5 Yorktown. I risultati hanno confermato l'elevata precisione ed efficacia del loro metodo di inclusione quantistica, stabilire un trampolino di lancio per risolvere molti diversi tipi di problemi di scienza dei materiali su un computer quantistico.
Galli ha osservato che, "Con l'inevitabile maturità dei computer quantistici, prevediamo che il nostro approccio sarà applicabile alla simulazione di regioni di interesse in molecole e materiali per la comprensione e la scoperta di catalizzatori e nuovi farmaci, così come soluzioni acquose contenenti specie complesse disciolte."
Il team di Galli fa parte del MICCoM, con sede ad Argonne; il Chicago Quantum Exchange, con sede a UChicago; e il progetto QISpin finanziato dall'Air Force Office of Scientific Research.
La loro ricerca ha sfruttato il software WEST sviluppato all'interno del MICCoM e ha fatto uso di diverse risorse informatiche oltre al computer quantistico IBM disponibile pubblicamente:l'Argonne Leadership Computing Facility e il National Energy Research Scientific Computing Center, entrambe le strutture per gli utenti dell'Ufficio delle scienze del DOE; e il Research Computing Center dell'Università di Chicago.
Il lavoro del team è presentato in un articolo intitolato "Quantum Simulations of Materials on Near-term Quantum Computer" che appare nel numero di luglio 2020 di npj Materiali di calcolo .