Proprio come gli esseri umani che li hanno creati, i computer trovano difficile la fisica, ma la meccanica quantistica ancora più difficile. Ma una nuova tecnica creata da tre scienziati dell'Università di Chicago consente ai computer di simulare determinati effetti quantomeccanici complessi in materiali elettronici complessi con uno sforzo molto minore.
Rendendo queste simulazioni più accurate ed efficienti, gli scienziati sperano che la tecnica possa aiutare a scoprire nuove molecole e materiali, come nuovi tipi di celle solari o computer quantistici.
"Questo progresso ha un immenso potenziale per approfondire la nostra comprensione dei fenomeni molecolari, con implicazioni significative per la chimica, la scienza dei materiali e i campi correlati", ha affermato lo scienziato Daniel Gibney, Ph.D. dell'Università di Chicago. studente di chimica e primo autore dell'articolo, pubblicato il 14 dicembre in Physical Review Letters .
Una foglia o un pannello solare sembrano lisci e semplici dall'esterno, ma ingrandisci il livello molecolare e vedrai una danza incredibilmente complicata di elettroni e molecole.
Per fare nuovi progressi nella sostenibilità, nella produzione, nell’agricoltura e in molti altri campi, gli scienziati modellano il comportamento di queste interazioni chimiche e molecolari. Ciò aiuta a rivelare nuove possibilità di progettazione per il futuro, per qualsiasi cosa, dai nuovi modi per sequestrare l'anidride carbonica a nuovi tipi di bit quantistici.
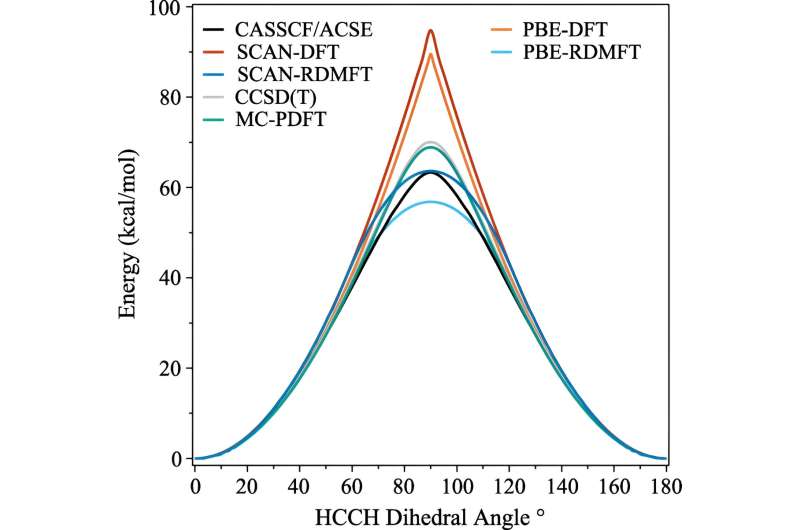
Negli ultimi decenni sono stati fatti molti passi avanti, ma una delle aree che è rimasta ostinatamente difficile da simulare è quando le molecole iniziano a mostrare comportamenti quantomeccanici complessi che gli scienziati chiamano correlazione forte.
Il problema è che una volta che gli elettroni iniziano a manifestare i loro effetti più quantomeccanici, come ad esempio diventare “impigliati”, i calcoli necessitano immediatamente di molta più potenza di calcolo. Anche i supercomputer faticano a gestire le implicazioni.
Uno dei calcoli comunemente usati è chiamato teoria del funzionale della densità. "Questa è fondamentalmente la tecnica più onnipresente per prevedere la struttura elettronica, ma è essenzialmente un'approssimazione in cui tutti gli elettroni sono trattati come una funzione di un elettrone", ha spiegato David Mazziotti, professore di chimica e autore senior dello studio.
Per molti calcoli, un'approssimazione è sufficiente. Ma inizia a crollare man mano che il comportamento degli elettroni diventa più correlato, come accade quando la meccanica quantistica inizia a entrare in gioco. Nella meccanica quantistica, questi elettroni possono trovarsi in più posti, o orbitali, contemporaneamente. Ciò ostacola non solo il cervello umano, ma anche la teoria del funzionale della densità.
"E questo è un problema importante, perché molte delle questioni che ci stanno a cuore nel 21° secolo, come le nuove molecole e i materiali per l'energia rinnovabile e la sostenibilità, ci impongono di sfruttare la natura quantistica dei materiali", ha affermato Mazziotti.
Mazziotti, Gibney e il terzo autore Jan-Niklas Boyn hanno scoperto che potrebbero aggiungere una correzione universale alla teoria del funzionale della densità che consente agli elettroni di rimanere impigliati tra più orbitali contemporaneamente.
"Ciò consente agli orbitali nel calcolo di essere non solo completamente pieni o completamente vuoti, ma ovunque nel mezzo", ha affermato Mazziotti. "Arriviamo a un'immagine a un elettrone che è ancora in grado di catturare il comportamento che deriva dagli effetti correlati degli elettroni a molti corpi."
Come bonus, hanno detto gli scienziati, il codice può essere aggiunto agli algoritmi esistenti senza doverlo riscrivere. "Fondamentalmente, la correzione interviene ogni volta che è necessaria, ma altrimenti non interferisce con il resto del codice", ha affermato Gibney.
È anche universale, in quanto può essere aggiunto al codice che simula molti tipi di comportamento elettronico, che si tratti di pannelli solari fotovoltaici, sequestro del carbonio o materiali superconduttori, o persino biologia.
Ad esempio, ha spiegato Boyn, un'applicazione potrebbe essere quella di comprendere la chimica che avviene utilizzando enzimi contenenti atomi metallici, noti come metalloenzimi.
"C'è una miriade di metalloenzimi responsabili di gran parte della chimica delle cellule, per esempio, ma sono notoriamente difficili da descrivere con i modelli attuali", ha detto. "Questa teoria potrebbe permetterci nel prossimo futuro di affrontare questa chimica in un modo che al momento è impossibile."
Ulteriori informazioni: Daniel Gibney et al, Generalizzazione universale della teoria del funzionale della densità per la correlazione statica, Lettere di revisione fisica (2023). DOI:10.1103/PhysRevLett.131.243003
Informazioni sul giornale: Lettere di revisione fisica
Fornito dall'Università di Chicago