Stuart Lindsay è il direttore del Center for Single Molecule Biophysics presso il Biodesign Institute dell'Arizona Arizona State University. Credito:The Biodesign Institute presso l'Arizona State University
Circa tre miliardi di coppie di basi costituiscono il genoma umano, la planimetria della vita. Nel 2003, il Progetto Genoma Umano ha annunciato il successo della decrittazione di questo codice, un tour de force che continua a fornire un flusso di intuizioni rilevanti per la salute e le malattie umane.
Tuttavia, gli attori primari in quasi tutti i processi vitali sono le proteine codificate da sequenze di DNA note come geni. Per un ampio spettro di malattie, le proteine possono fornire rivelazioni molto più convincenti di quelle che possono essere raccolte dal solo DNA, se i ricercatori riescono a sbloccare le sequenze di amminoacidi da cui sono composti.
Ora, Stuart Lindsay e i suoi colleghi del Biodesign Institute dell'Arizona State University hanno compiuto un passo importante in questa direzione, dimostrando l'accurata identificazione degli amminoacidi, fissando brevemente ciascuno in una stretta giunzione tra una coppia di elettrodi fiancheggianti e misurando una catena caratteristica di picchi di corrente che passano attraverso successive molecole di amminoacidi.
Utilizzando un algoritmo di apprendimento automatico, Lindsay e il suo team sono stati in grado di addestrare un computer a riconoscere esplosioni di attività elettrica che rappresentano il legame momentaneo di un amminoacido all'interno della giunzione. I segnali di rumore hanno dimostrato di agire come impronte digitali affidabili, identificazione degli amminoacidi, comprese varianti leggermente modificate.
Le proteine stanno già fornendo una grande quantità di informazioni relative a malattie tra cui il cancro, diabete e disturbi neurologici come l'Alzheimer, oltre a fornire informazioni chiave su un altro processo mediato dalle proteine:l'invecchiamento.
Il nuovo lavoro fa avanzare la prospettiva del sequenziamento clinico delle proteine e la scoperta di nuovi biomarcatori, segnali di allerta precoce che segnalano la malattia. Ulteriore, il sequenziamento delle proteine può trasformare radicalmente il trattamento del paziente, consentendo un monitoraggio preciso della risposta della malattia alle terapie, a livello molecolare.
I risultati della ricerca del gruppo sono riportati nell'edizione online avanzata della rivista Nanotecnologia della natura .
Dal genoma al proteoma
Un'enorme libreria di proteine, nota come proteoma, occupa il centro della scena in quasi tutti i processi vitali. Le proteine sono vitali per la crescita cellulare, differenziazione e riparazione; catalizzano reazioni chimiche e forniscono difesa contro le malattie, tra una miriade di funzioni domestiche.
Una delle sorprese più strane emerse dallo Human Genome Project è il fatto che solo l'1,5% circa del genoma codifica per le proteine. Il resto dei nucleotidi del DNA forma sequenze regolatorie, geni RNA non codificanti, introni, e DNA non codificante, (una volta etichettato in modo derisorio "DNA spazzatura"). Questo lascia gli umani con uno scarso 20-25, 000 geni, una scoperta che fa riflettere, dato che l'umile nematode ha all'incirca lo stesso numero. Come osserva il professor Lindsay, la notizia peggiora:"Una pianta di giglio ha circa un ordine di grandezza in più di geni di noi, " lui dice.
Il mistero di organismi complessi come gli esseri umani che portano un numero di geni spaventosamente basso ha a che fare con il fatto che le proteine generate dal progetto del DNA possono essere modificate in diversi modi. Infatti, gli scienziati hanno già identificato oltre 100, 000 proteine umane e ricercatori come Lindsay credono che questa possa essere solo la punta dell'iceberg.
Proprio come le frasi possono avere il loro significato alterato attraverso cambiamenti nell'ordine delle parole o nella punteggiatura, le proteine generate da modelli genici possono cambiare funzione (o talvolta essere rese inutilizzabili), spesso con gravi conseguenze per la salute umana. Due processi chiave che modificano le proteine sono noti come splicing alternativo e modifica post-traduzionale. Sono loro i driver della straordinaria variazione proteica osservata.
Lo splicing alternativo si verifica quando si codificano regioni di RNA, (noti come esoni) vengono uniti insieme e le regioni non codificanti (noti come introni) vengono tagliate fuori, prima della traduzione in proteine. Questo processo non avviene sempre in modo ordinato, con l'introduzione di occasionali sovrapposizioni di esoni o introni, produzione di proteine splicing alternativo, la cui funzione può essere alterata.
Le modifiche post-traduzionali sono marcatori aggiunti dopo che le proteine sono state prodotte. Esistono molte forme di modifica post-traduzionale, comprese metilazione e fosforilazione. Alcune proteine alterate svolgono funzioni vitali, mentre altri possono essere aberranti e associati a malattie (o propensione alla malattia). Un certo numero di tumori sono associati a tali errori proteici, che sono già utilizzati come marker diagnostici. La corretta identificazione di tali proteine rimane tuttavia una grande sfida in biomedicina.
Nuove sequenze
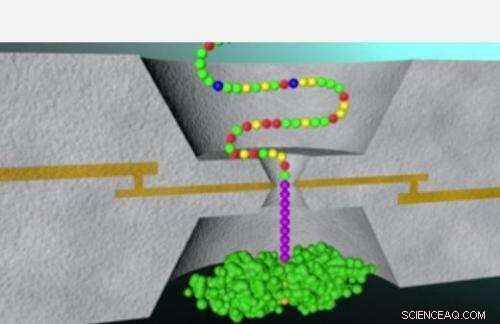
La tecnica descritta nella ricerca attuale è stata precedentemente applicata nel laboratorio di Lindsay per il sequenziamento riuscito delle basi del DNA. Questo metodo, noto come tunneling di riconoscimento, prevede l'infilatura di un peptide attraverso un minuscolo occhiello noto come nanoporo. Un paio di elettrodi metallici, separati da uno spazio di circa due nanometri, si trova su entrambi i lati del nanoporo mentre unità successive di un peptide vengono infilate attraverso la minuscola apertura, con ogni unità che completa un circuito elettrico ed emette una raffica di picchi di corrente.
Il gruppo di ricerca ha dimostrato che analisi ravvicinate di questi picchi di corrente potrebbero consentire ai ricercatori di determinare quale delle quattro basi nucleotidiche:adenina, timina, citosina o guanina - era in bilico tra gli elettrodi nel nanoporo.
"Circa 2 anni fa in uno dei nostri incontri di laboratorio, è stato suggerito che forse la stessa tecnologia avrebbe funzionato per gli amminoacidi, "Dice Lindsay. Così sono iniziati gli sforzi per affrontare la sfida sostanzialmente più grande dell'utilizzo del tunneling di riconoscimento per identificare tutti i 20 amminoacidi presenti nelle proteine, al contrario delle sole 4 basi che comprendono il DNA.
Il sequenziamento di singole molecole delle proteine è di enorme valore, offering the potential to detect diminishingly small quantities of proteins that may have been tweaked by alternative splicing or post-translational modification. Spesso, these are the very proteins of interest from the standpoint of recognizing disease states, though current technologies are inadequate to detect them.
As Lindsay notes, there is no equivalent in the protein world to polymerase chain reaction (PCR) technology, which allows minute quantities of DNA in a sample to be rapidly amplified. "We probably don't even know about most of the proteins that would be important in diagnostics. It's just a black hole to us because the concentrations are too low for current analytical techniques, " lui dice, adding that the ability of recognition tunneling to pinpoint abnormalities on a single molecule basis "could be a complete game changer in proteomics."
The new paper describes a series of experiments in which pure samples of individual amino acids, individual molecules in mixed solution and finally, short peptide chains were successfully identified through recognition tunneling. The work sets the stage for a method to sequence individual protein molecules rapidly and cheaply (see accompanying animation).
A machine learning algorithm known as Support Vector Machine was used to train a computer to analyze the burst signals produced when amino acids formed bonds in the tunnel junction and emitted a lively noise signal as the poised electrodes passed tunneling current through each molecule. (The machine learning algorithm is the same one used by the IBM computer 'Watson' to defeat a human opponent in Jeopardy.)
Lindsay says that around 50 distinct signal burst characteristics were used in the amino acid identifications, but that most of the discriminatory power is achieved with 10 or fewer signal traits.
Remarkably, recognition tunneling not only pinpointed amino acids with high reliability from single complex burst signals, but managed to distinguish a post-translationally modified protein (sarcosine) from its unmodified precursor (glycine) and also to discriminate between mirror-image molecules knows as enantiomers and so-called isobaric molecules, which differ in peptide sequence but exhibit identical masses.
Pathway to the $1000 dollar proteome?
Lindsay indicates that the new studies, which rely on innovative strategies for handling single molecules coupled with startling advances in computing power, open up horizons that were inconceivable only a short time ago. It is becoming clear that the tools that made the $1000 genome feasible are equally applicable to an eventual $1000 dollar proteome. Infatti, such a landmark may not be far off. "Perchè no?" Lindsay asks. "People think it's crazy but the technical tools are there and what will work for DNA sequencing will work for protein sequencing."
While the tunneling measurements have until now been made using a complex laboratory instrument known as a scanning tunneling microscope (STM), Lindsay and his colleagues are currently working on a solid state device capable of fast, cost-effective and clinically applicable recognition tunneling of amino acids and other analytes. Eventual application of such solid-state devices in massively parallel systems should make clinical proteomics a practical reality.