
Membri CAMERA (da sinistra) Peter Zwart, Jeff Donatelli e Kanupriya Pande, coautori di un articolo che descrive come l'algoritmo M-TIP del gruppo ha determinato le strutture virali 3D dai dati di diffrazione di singole particelle. Donatelli tiene in mano un modello stampato in 3D di uno dei virus ricostruiti da M-TIP. Credito:Marilyn Chung, Berkeley Lab
Come parte di un gruppo di ricerca internazionale, Jeff Donatelli, Peter Zwart e Kanupriya Pande del Center for Advanced Mathematics for Energy Research Applications (CAMERA) presso il Lawrence Berkeley National Laboratory (Berkeley Lab) hanno contribuito con algoritmi chiave che hanno contribuito a raggiungere un obiettivo proposto per la prima volta più di 40 anni fa, utilizzando le correlazioni angolari dei raggi X istantanee da molecole non cristalline per determinare la struttura 3D di importanti oggetti biologici. Questa tecnica ha il potenziale per consentire agli scienziati di far luce su strutture e dinamiche biologiche che in precedenza erano impossibili da osservare con i tradizionali metodi a raggi X.
La svolta è stata il risultato di un esperimento di diffrazione a particella singola condotto presso la Linac Coherent Light Source (LCLS) del Dipartimento dell'Energia (DOE) dall'iniziativa Single-Particle organizzata dallo SLAC National Accelerator Laboratory. Nell'ambito di questa iniziativa, il team CAMERA ha unito gli sforzi con Ruslan Kurta, un fisico presso la struttura europea XFEL (laser a elettroni liberi a raggi X) in Germania, analizzare le correlazioni angolari dai dati sperimentali e utilizzare l'algoritmo di fase iterativa multilivello (M-TIP) di CAMERA per eseguire le prime ricostruzioni virali 3D di successo da correlazioni sperimentali. I risultati sono stati descritti in un articolo pubblicato il 12 ottobre in Lettere di revisione fisica .
"Negli ultimi 40 anni, questo era considerato un problema che non poteva essere risolto, " ha detto Peter Zwart, co-autore dell'articolo e un bioscienziato fisico che è membro di CAMERA con sede presso la divisione di biofisica molecolare e imaging integrato presso il Berkeley Lab. "Ma si scopre che gli strumenti matematici che abbiamo sviluppato sono in grado di sfruttare le informazioni aggiuntive nascoste nel problema che era stato precedentemente trascurato. È gratificante vedere che il nostro approccio teorico porta a uno strumento pratico".
Nuove opportunità di ricerca abilitate da XFELs
Per gran parte del secolo scorso, la tecnica di riferimento per determinare la struttura molecolare ad alta risoluzione è stata la cristallografia a raggi X, dove il campione di interesse è disposto in un ampio reticolo periodico ed esposto a raggi X che si disperdono e formano schemi di diffrazione che vengono raccolti su un rivelatore. Anche se la cristallografia è riuscita a determinare molte strutture ad alta risoluzione, è difficile utilizzare questa tecnica per studiare strutture che non sono suscettibili di cristallizzazione o cambiamenti strutturali che non si verificano naturalmente all'interno di un cristallo.
La creazione di strutture XFEL, tra cui Linac Coherent Light Source (LCLS) e European X-FEL, hanno creato opportunità per condurre nuovi esperimenti in grado di superare i limiti della cristallografia tradizionale. In particolare, I fasci XFEL sono diversi ordini di grandezza più luminosi e hanno lunghezze di impulso molto più brevi rispetto alle tradizionali sorgenti di luce a raggi X, che consentono loro di raccogliere segnali di diffrazione misurabili da campioni non cristallizzati più piccoli e anche di studiare le dinamiche veloci. La diffrazione di una singola particella è una di queste tecniche sperimentali emergenti abilitate da XFELS, dove si raccolgono immagini di diffrazione da singole molecole invece che da cristalli. Queste tecniche a particella singola possono essere utilizzate per studiare la struttura molecolare e le dinamiche che sono state difficili da studiare con le tecniche di imaging tradizionali.
Superare i limiti nell'imaging a particella singola tramite correlazioni angolari
Una delle principali sfide dell'imaging a particella singola è quella della determinazione dell'orientamento. "In un esperimento su una singola particella, non hai il controllo sulla rotazione delle particelle poiché vengono colpite dal raggio di raggi X, quindi ogni istantanea di un successo conterrà informazioni sul campione da un orientamento diverso, " ha detto il co-autore Jeff Donatelli, un matematico applicato in CAMERA che ha sviluppato molti degli algoritmi nel nuovo framework. "La maggior parte degli approcci all'analisi delle singole particelle si è finora basata sul tentativo di determinare questi orientamenti delle particelle dalle immagini; tuttavia, la migliore risoluzione ottenibile da queste analisi è limitata dalla precisione con cui questi orientamenti possono essere determinati da dati rumorosi."
Invece di cercare di determinare direttamente questi orientamenti, il team ha adottato un approccio diverso basato su un'idea originariamente proposta negli anni '70 da Zvi Kam. "Piuttosto che esaminare le singole intensità dei dati nel tentativo di trovare l'orientamento corretto per ogni fotogramma misurato, eliminiamo del tutto questo passaggio utilizzando le cosiddette funzioni di cross-correlazione, " disse Kurta.
Questo approccio, noto come diffusione di raggi X di fluttuazione, si basa sull'analisi delle correlazioni angolari di ultracorti, intensi impulsi di raggi X diffusi da biomolecole non cristalline. "La bellezza dell'utilizzo dei dati di correlazione è che contengono un'impronta digitale completa della struttura 3D di un oggetto che estende gli approcci tradizionali di dispersione delle soluzioni, " Ha detto Zwart.
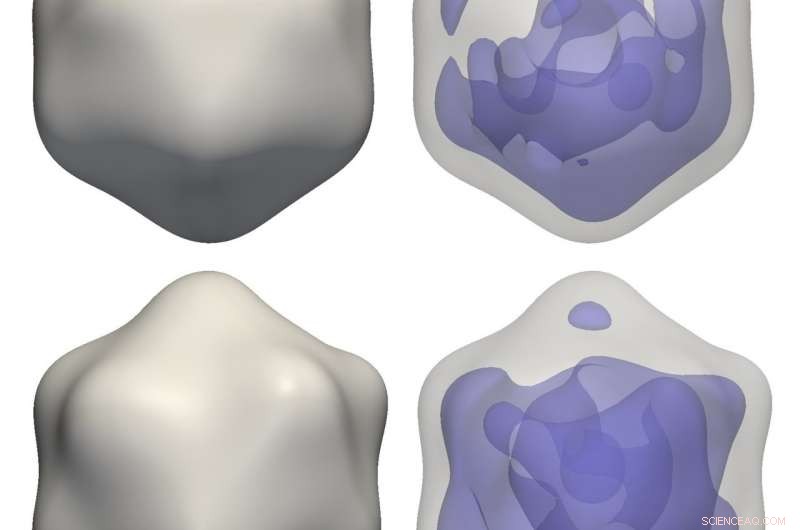
Virus ricostruiti:ricostruzioni di un virus nano del riso (in alto) e di un batteriofago PR772 (in basso) da dati di correlazione sperimentale utilizzando M-TIP. Le immagini a destra mostrano le asimmetrie nel materiale genetico interno per ogni ricostruzione del virus. Credito:Jeff Donatelli, Berkeley Lab
Ricostruire la struttura 3D dalle correlazioni con l'algoritmo M-TIP di CAMERA
La svolta del team nella ricostruzione della struttura 3D dai dati di correlazione è stata resa possibile dall'algoritmo di fase iterativa multilivello (M-TIP) sviluppato da CAMERA. "Tra i principali vantaggi di M-TIP c'è la sua capacità di risolvere la struttura direttamente dai dati di correlazione senza dover fare affidamento su vincoli di simmetria, e, ma ancora più importante, senza la necessità di risolvere il problema della determinazione dell'orientamento, "Ha detto Donatelli.
Donatelli, Il direttore di CAMERA James Sethian e Zwart hanno sviluppato il loro framework M-TIP sviluppando una generalizzazione matematica di una classe di algoritmi noti come tecniche di phasing iterative, che vengono utilizzati per determinare la struttura in un problema più semplice, noto come recupero di fase. Un documento che descrive il framework M-TIP originale è stato pubblicato nell'agosto 2015 nel Atti dell'Accademia Nazionale delle Scienze .
"Analisi di correlazione avanzate in combinazione con ricostruzioni ab-initio di M-TIP definiscono chiaramente un percorso efficiente per l'analisi strutturale di oggetti su scala nanometrica a XFELs, " Ha detto Zwart.
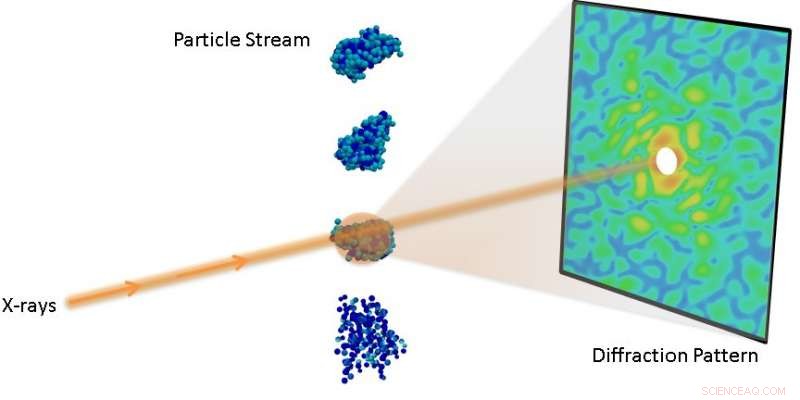
Configurazione sperimentale per un esperimento di diffrazione di una singola particella. Credito:Lawrence Berkeley National Laboratory
Direzioni future per l'analisi della correlazione e M-TIP
Il team osserva che i metodi utilizzati in questa analisi possono essere applicati anche per analizzare i dati di diffrazione quando c'è più di una particella per colpo.
"La maggior parte degli algoritmi per l'imaging a particella singola può gestire solo una molecola alla volta, limitando così segnale e risoluzione. Il nostro approccio, d'altra parte, è scalabile in modo che dovremmo anche essere in grado di misurare più di una particella alla volta, " ha affermato Kurta. L'imaging con più di una particella per colpo consentirà agli scienziati di ottenere percentuali di successo molto più elevate, poiché è più facile usare un raggio ampio e colpire molte particelle alla volta, ed eviterà anche la necessità di separare i colpi di una singola particella dai colpi di più particelle e dai colpi a salve, che è un altro requisito impegnativo nell'imaging a particella singola tradizionale.
Come parte della suite di strumenti di calcolo di CAMERA, hanno anche sviluppato una versione diversa di M-TIP che risolve il problema dell'orientamento e può classificare le immagini in stati conformazionali, e di conseguenza può essere utilizzato per studiare piccole differenze biologiche nel campione misurato. Questa versione alternativa di M-TIP è stata descritta in un documento pubblicato il 26 giugno 2017 nel Atti dell'Accademia Nazionale delle Scienze e fa parte di una nuova iniziativa di collaborazione tra SLAC National Accelerator Laboratory, TELECAMERA, the National Energy Research Scientific Computing Center (NERSC) and Los Alamos National Laboratory as part of DOE's Exascale Computing Project (ECP).














