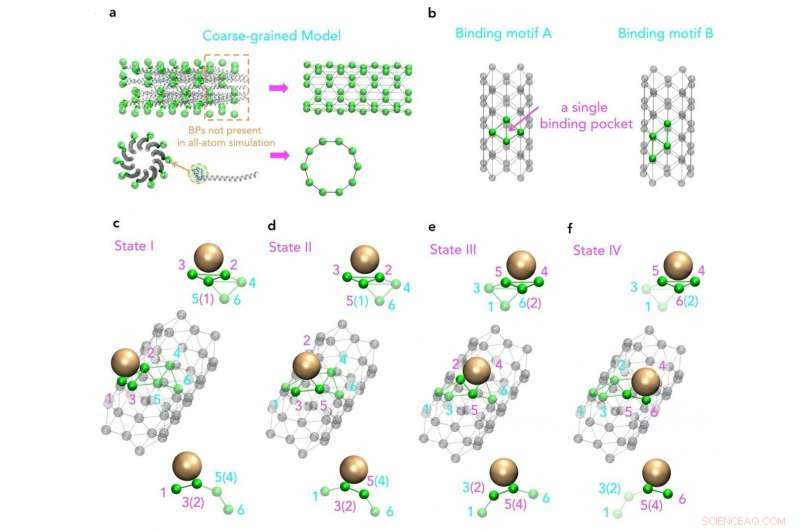
Vista molecolare di un modello a grana grossa basato sulla struttura originale delle principali proteine di rivestimento M13 Credito:SUTD
Le simulazioni atomistiche sono un potente strumento per studiare il movimento e le interazioni di atomi e molecole. In molti processi biologici, effetti su larga scala, Per esempio, l'assemblaggio di virus di grandi dimensioni su nanoparticelle è importante. I processi di assemblaggio di questi grandi virus sono di fondamentale importanza per la progettazione di molti dispositivi e terapie mirate alle proteine virali. Però, la scala temporale e di lunghezza di questi processi di assemblaggio è solitamente troppo grande per le simulazioni a risoluzione molecolare.
Inoltre, anche se un aumento della potenza di calcolo consente simulazioni più complesse e più lunghe, strutture virali, come M13, sono ancora fuori portata. Ecco perché un gruppo di ricerca della Singapore University of Technology and Design (SUTD) e del Massachusetts Institute of Technology (MIT) ha sviluppato una procedura che collega i processi di assemblaggio su larga scala alle simulazioni molecolari. Assistente Prof Desmond Loke della SUTD's Science, Il cluster di matematica e tecnologia ha affermato:"Per la simulazione di M13, abbiamo iniziato con diversi set di campi di forza. Sono stati scelti campi di forza adatti e sono stati utilizzati come input per simulazioni di dinamiche molecolari con il modello a grana grossa progettato per catturare il modello chiave del processo di assemblaggio".
"Mentre sappiamo che la produzione basata su M13 può essere fondamentalmente guidata da interazioni nanoparticelle-peptidi, che può anche essere un principio chiave alla base della bioingegneria di tipo M13, abbiamo poca conoscenza di come pattern ripetuti di peptidi a estremità corta su una superficie M13 siano effettivamente coinvolti in queste interazioni. Per studiare questo, idealmente dobbiamo includere una struttura completa della proteina di rivestimento virale, che è un compito difficile per le attuali simulazioni di dinamica molecolare all'avanguardia, " aggiunge la Dott.ssa Lunna Li, primo autore dell'articolo.
La procedura consente agli utenti di aggiungere diversi tipi di nanoparticelle a una soluzione, a un livello realistico. Ispirato da questa procedura, L'assistente professore Loke ei suoi colleghi sono stati in grado di simulare un virus su larga scala con nanoparticelle e all'interno di una soluzione per cinquanta nanosecondi.
Il dottor Li ha detto, "La struttura del virus e la soluzione contengono circa 700, 000 atomi in totale." Considerando la forma e le dimensioni delle caratteristiche, la complessità di questa simulazione può essere maggiore di qualsiasi simulazione eseguita in precedenza.
"Una simulazione eseguita in microsecondi sarebbe stata possibile se fosse stato utilizzato un modello M13 più piccolo, ma può essere utile ridurre il tempo per osservare effettivamente come l'intera struttura può influenzare l'assemblaggio tra M13 e le nanoparticelle, " ha spiegato l'assistente Prof Loke.