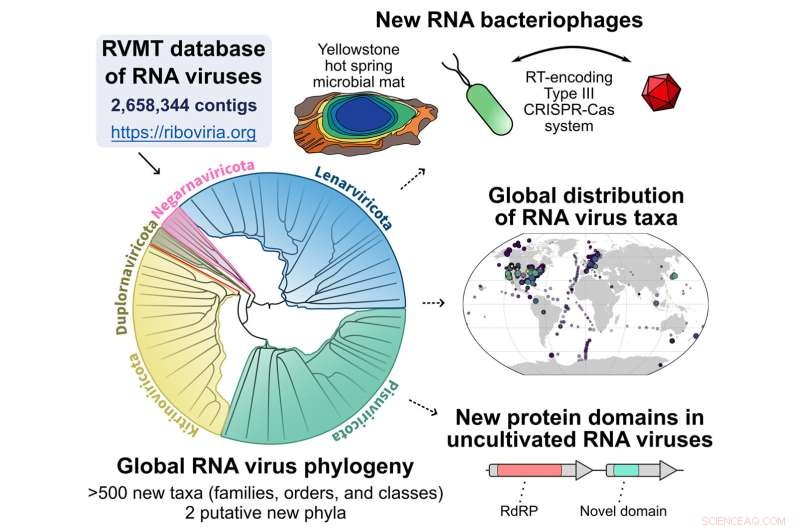
Panoramica grafica della pipeline a partire dal database RNA Virus MetaTranscriptomes (RVMT) per scoprire l'espansione nella diversità dei virus RNA. Credito:Simon Roux
Uno zoo una volta offriva un libro da colorare con gli orsi polari nelle scene invernali che veniva fornito con pastelli in varie sfumature di bianco. Per i ricercatori che cercano sequenze di virus a RNA in grandi set di dati, il loro lavoro potrebbe essere simile a trovare un singolo fiocco di neve su una pagina colorata di quel libro.
Pubblicato online il 28 settembre 2022 in Cell , un team guidato da ricercatori dell'Università di Tel Aviv in Israele, del National Center for Biotechnology Information e del Joint Genome Institute (JGI) del Dipartimento dell'Energia degli Stati Uniti (DOE), una struttura per gli utenti dell'Office of Science del DOE situata presso il Lawrence Berkeley National Laboratory ( Berkeley Lab) descrivono una pipeline computazionale in grado di scansionare in modo specifico quei fiocchi di neve o sequenze di virus a RNA. Utilizzando questo flusso di lavoro, il team ha analizzato oltre 5.000 set di dati di sequenze di RNA (metatrascrittomi) generati da diversi campioni ambientali in tutto il mondo, determinando un aumento di cinque volte della diversità dei virus dell'RNA.
"Il mondo dei virus che ci circonda è vasto e ora abbiamo i mezzi per esplorarlo", ha affermato Eugene Koonin, ricercatore senior presso l'NCBI e uno degli autori senior del documento, della diversità virale scoperta. "Sebbene le sfide tecniche dell'analisi dei dati su questa scala siano formidabili."
Setacci computazionali per filtrare le sequenze
Ci sono più microbi sul pianeta che particelle in una manciata di sporco e i virus superano di gran lunga i microbi. I progressi nelle tecnologie di sequenziamento e negli strumenti computazionali hanno scoperto una varietà di virus che infettano non solo le colture, gli animali e gli esseri umani, ma anche i microbi la cui presenza o assenza può influire sui cicli dei nutrienti del pianeta.
Mentre la maggior parte delle informazioni genetiche dell'organismo è codificata nel DNA, con l'RNA che fornisce le istruzioni all'interno del DNA alla cellula, i virus a RNA memorizzano le loro informazioni genetiche nell'RNA senza uno stadio del DNA. "Direi che i virus a RNA a livello globale sono ancora meno conosciuti dei virus a DNA", ha affermato Simon Roux, uno scienziato del JGI e uno dei co-responsabili del progetto. "Ma come i virus a DNA, i virus a RNA infettano i microbi in tutto il mondo e portano alla morte cellulare e/o a profondi cambiamenti nella fisiologia cellulare durante l'infezione".
Sebbene tutti i virus a RNA abbiano un gene che codifica per un enzima chiamato RNA polimerasi (RdRP) diretto da RNS, necessario per replicare la replicazione del genoma dell'RNA, rilevarlo è stata una sfida. Trovare i fiocchi di neve del virus RNA nella tempesta di neve dei dati genomici ha comportato lo sviluppo di speciali setacci computazionali per filtrare le sequenze che difficilmente contenessero la sequenza RdRP.
Il lavoro è il risultato di una collaborazione a tre vie iniziata nel 2019, ha ricordato Uri Neri dell'Università di Tel Aviv, uno dei co-responsabili del progetto e primo autore dello studio. I membri dei team di Tel Aviv e NCBI, che stavano già lavorando insieme all'estrazione di virus procariotici, hanno appreso da Nikos Kyrpides di JGI che il suo gruppo Microbiome Data Science stava lavorando anche all'estrazione di virus RNA. Dopo un paio di riunioni virtuali dei tre team, è stato chiaro che uno sforzo collaborativo più ampio sarebbe stato molto più efficace per ottenere risultati di qualità superiore rispetto a sforzi individuali minori. Questo è anche il tipo di spirito di comunità sinergico e collaborativo che il JGI sostiene e promuove attivamente.
Il team ha utilizzato tutti i set di dati sul metatrascrittoma pubblicamente disponibili dal sistema Integrated Microbial Genomes &Microbiomes (IMG/M) del JGI. "Abbiamo quindi esaminato molti altri campioni e perfezionato la nostra metodologia", ha affermato Neri. "Il nostro team è cresciuto, così come l'ambito del progetto". A tal fine, ha sottolineato Kyrpides, i contributi dei numerosi utenti scientifici della JGI nella raccolta e nella presentazione dei loro campioni di microbioma per il sequenziamento presso la JGI non possono essere sopravvalutati. La loro cooperazione e supporto, ha affermato, e in molti casi il loro permesso di utilizzare dati di sequenza non ancora pubblicati, sono stati assolutamente fondamentali per il successo di questo sforzo, così come il riconoscimento del loro contributo.
Sia Roux che Koonin hanno notato che la pletora di sequenze di virus a RNA scoperte "cambia in modo significativo la visione globale della diversità dei virus", sebbene non nelle classificazioni di livello superiore dei gruppi di virus (phyla). Le nuove sequenze stanno colmando alcune lacune sui virus esistenti gruppi aggiungendo anche nuovi rami. Inoltre, i virus a RNA non sembrano essere distribuiti uniformemente nel mondo.
Un gruppo allargato è di virus associati a batteri; fino ad ora, la maggior parte dei virus a RNA conosciuti sono stati associati agli eucarioti. Insieme all'espansione dei virus a RNA associati ai batteri c'è la scoperta che "alcuni batteri usano CRISPR per difendersi dall'RNA", ha osservato Roux, "sebbene non sia chiaro il motivo per cui questo viene rilevato così raramente".
Sviluppo di approcci per conciliare i Big Data "reali"
Per il team, il lavoro computazionale che ha portato alla scoperta dell'abbondanza di virus a RNA è solo l'inizio. "Dico spesso che identificare una sequenza come virale non è nemmeno metà della storia". ha detto Neri. "Abbiamo investito molti dei nostri sforzi nelle analisi post-scoperta:nel miglior modo possibile, abbiamo cercato di descrivere i domini proteici trasportati da ogni virus e chi è il loro probabile ospite. Abbiamo reso tutte queste informazioni completamente gratuite e apertamente a disposizione della più ampia comunità scientifica."
Uri Gophna dell'Università di Tel Aviv e Koonin hanno entrambi notato che altre ricerche parallele hanno riportato simili "espansioni drammatiche" del viroma globale dell'RNA. "Ora dobbiamo confrontare e riconciliare i risultati, arrivando a un unico set di dati non ridondante", ha affermato Koonin. "Speriamo che in tempi relativamente brevi saremo in grado di stimare la dimensione effettiva del viroma dell'RNA. Tuttavia, ora si tratta di veri Big Data, abbiamo a che fare con miliardi di sequenze e, presto, con trilioni. Lo sviluppo di approcci efficienti e automatizzati per l'analisi e classificare i dati di sequenza su questa scala è essenziale." + Esplora ulteriormente