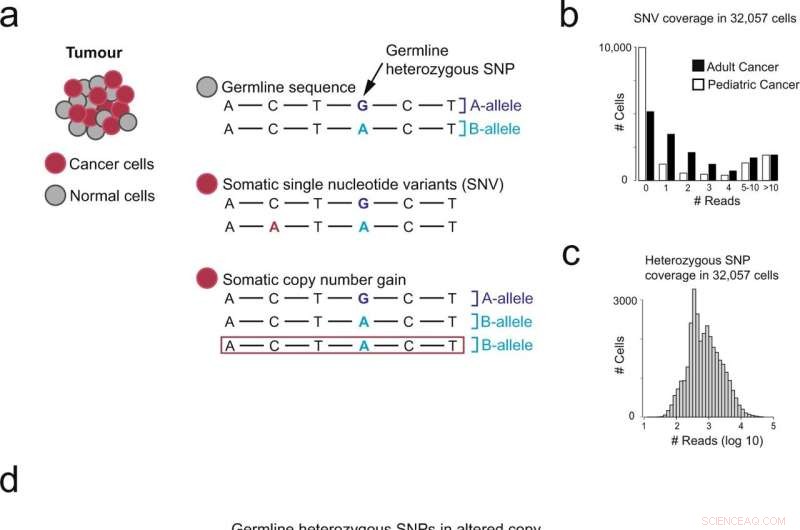
Panoramica di diversi approcci per identificare le cellule derivate dal cancro. a Cambiamenti genomici presenti nei genomi del cancro. b Numero di cellule (asse y) con letture N che coprono mutazioni puntiformi (asse x), separate da un carico di mutazioni basso (neuroblastoma NB) e alto (carcinoma a cellule renali RCC). c Numero di cellule (asse y) con letture N che coprono polimorfismi eterozigoti a singolo nucleotide (SNP) (asse x). d Panoramica sull'utilizzo di spostamenti allelici che rappresentano le modifiche del numero di copie per rilevare le cellule tumorali. Credito:Biologia delle comunicazioni (2022). DOI:10.1038/s42003-022-03808-9. https://www.nature.com/articles/s42003-022-03808-9
Un nuovo metodo per separare le cellule tumorali dalle cellule non cancerose è stato sviluppato dai ricercatori del Wellcome Sanger Institute, per dare una spinta a coloro che lavorano per comprendere meglio la biologia del cancro utilizzando il sequenziamento dell'mRNA unicellulare.
Lo studio, pubblicato oggi su Biologia delle comunicazioni , migliora i metodi esistenti per identificare quali cellule in un campione sono cancerose e fornisce dati cruciali sul microambiente dei tumori. Il software è apertamente disponibile per i ricercatori di tutto il mondo da applicare ai propri dati, migliorando l'efficacia del sequenziamento unicellulare per comprendere il cancro.
L'analisi dell'mRNA unicellulare delle cellule tumorali è una delle tecniche all'avanguardia utilizzate per comprendere meglio la biologia del cancro. I dati generati possono essere utilizzati per cercare di distruggere i tumori con i farmaci o capire in primo luogo come si manifestano i tumori.
Un passaggio fondamentale in questo processo è la separazione delle cellule cancerose e non cancerose, ma questo non è sempre un compito facile. Oltre ai molti tipi di cancro, ci saranno anche differenze molecolari tra cellule tumorali dello stesso tipo all'interno di un singolo tumore.
Attualmente, il metodo migliore per farlo è misurare l'espressione genica media delle cellule nel campione, con un'espressione superiore o inferiore utilizzata per distinguere le cellule tumorali dalle cellule sane. Ma questo metodo può portare a false conclusioni.
In questo nuovo studio, i ricercatori del Wellcome Sanger Institute hanno eseguito il sequenziamento dell'intero genoma e il sequenziamento dell'mRNA unicellulare su campioni raccolti dal Great Ormond Street Hospital (GOSH).
Individuando gli squilibri degli alleli in questi dati, che indicano cambiamenti nel numero di copie nel genoma, il team è stato in grado di identificare le cellule tumorali in modo più affidabile rispetto ai metodi precedenti. Questo approccio sarà utile principalmente per convalidare nuovi tipi di cellule tumorali e comprendere meglio il microambiente del tessuto tumorale.
"Essere in grado di sapere in che modo il trascrittoma è diverso nelle cellule con genomi aberranti, come quelli che si trovano nei tumori, è una conoscenza preziosa e amplierà le domande a cui possiamo rispondere utilizzando il sequenziamento unicellulare", afferma il dott. Matt Young.
Il metodo, denominato alleleIntegrator, è disponibile come pacchetto software per l'utilizzo da parte dei ricercatori di tutto il mondo. + Esplora ulteriormente














