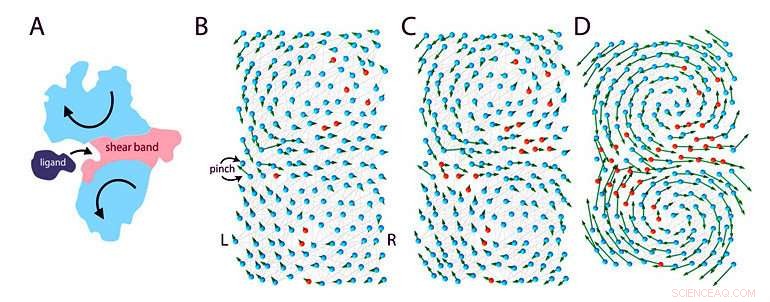
Figura 1:Modello elastico di una proteina che si lega a un ligando. (A) Quando una proteina si lega a un ligando, subisce un movimento su larga scala (frecce) che sono le firme delle proteine funzionali di piegatura. Ciò è possibile solo grazie alla presenza di alcune regioni "flaccide" ("banda di taglio" rosa) attraverso la proteina che separano le regioni rigide (blu) della proteina in due domini. (B)-(D) Il team ha modellato una proteina di 200 aminoacidi durante diverse fasi dell'evoluzione:passando da uno stato non funzionale (B) a uno funzionale (D). La proteina è modellata come una rete elastica elastica con due tipi di amminoacidi, modellati come perline:gli amminoacidi rosa sono flessibili e gli amminoacidi blu sono rigidi. I ricercatori imitano l'evoluzione cambiando un amminoacido casuale alla volta (mutazione) dal rosa al blu. Inizialmente, la proteina è per lo più rigida e non funzionale. Durante l'evoluzione, vengono aggiunti amminoacidi flessibili, alcuni utili, alcuni no. Col tempo, una regione "floscia" si forma al centro della molecola rendendo la proteina più flessibile per piegarsi e legarsi al ligando. Il modello ha stimato che si raggiunge una soluzione efficiente dopo mille mutazioni. Credito:Istituto per le scienze di base
Un team internazionale ha sviluppato un modello che simula l'evoluzione delle proteine. Partendo da rigido, proteine non funzionali, il modello al computer mostra come i componenti proteici in evoluzione possono lavorare insieme per dare origine a macchine molecolari dinamiche ed efficienti. La flessibilità consente alle proteine di modificare la loro conformazione 3D per legarsi ad altre molecole:questa proprietà è fondamentale per la loro funzione. Prof. Tsvi Tlusty e Dr. Sandipan Dutta presso il Center for Soft and Living Matter, all'interno dell'Istituto per le Scienze di Base (IBS, Corea del Sud), in collaborazione con il Prof. Albert Libchaber dell'Università Rockefeller e il Prof. Jean-Pierre Eckmann dell'Università di Ginevra hanno imitato l'evoluzione genica per ottenere proteine che possono piegarsi e legarsi ad altre molecole. La comprensione di questa relazione è uno degli aspetti più ricercati della biologia delle proteine; potrebbe aiutare a spiegare l'azione farmaceutica dei farmaci che si legano ai loro bersagli.
L'evoluzione ha plasmato il mondo vivente che vediamo intorno a noi per miliardi di anni. Miliardi di proteine lavorano in armonia per mantenere in corso questi processi vitali. Sono responsabili del buon funzionamento di qualsiasi organismo:riconoscono altre molecole (leganti), legarli a loro e convertirli. Altri hanno funzione di trasporto, fornire struttura, e sostegno alle cellule. I geni immagazzinano le informazioni sulla produzione e la progettazione di queste macchine molecolari. Però, nonostante decenni di ricerche, disegnare la "mappa" che disegna il percorso dai geni alla funzione proteica non è banale.
Secondo una recente ipotesi, la funzione proteica si basa su "articolazioni flessibili". Questo studio, pubblicato in Atti dell'Accademia Nazionale delle Scienze ( PNAS ), esamina il legame tra funzione e flessibilità modellando proteine come le reti elastiche. In questo modello, le proteine sono costituite da amminoacidi flessibili (polari) e rigidi (idrofobici) collegati da "molle" molecolari. Se alcune regioni della proteina sono sufficientemente flessibili, formano un canale "floppy", e l'intera macchina molecolare può piegarsi come un cardine. Questo movimento consente loro di legarsi efficacemente ad altre molecole. Il legame tra un ligando e una proteina rigida o flessibile può essere pensato come una palla che atterra su una roccia o su un cuscino morbido. È probabile che la palla rimbalzi via dopo aver colpito la roccia, ma è più probabile che il cuscino lo accetti. Perciò, la proteina flessibile è un legante migliore.
In questo modello, i geni immagazzinano i dettagli della struttura proteica in modo binario:gli amminoacidi flessibili sono immagazzinati come zeri e gli amminoacidi rigidi come uno. Di conseguenza, l'intera struttura proteica può essere semplificata come codice, come 11110001...111, simile alla memoria digitale di un computer. Però, non tutti i codici danno origine a proteine funzionali, ad esempio un codice con solo quelli:111111…1111, darebbe origine a una proteina completamente rigida, impossibilitato a muoversi, e non funzionale. Tra tutti i codici possibili, solo alcuni producono una proteina funzionale con al centro una regione "flaccida" che può accogliere il ligando.
Il modello imita l'evoluzione cambiando un amminoacido casuale alla volta. Durante l'evoluzione, gli zeri e gli uno nel gene vengono capovolti casualmente attraverso un processo chiamato mutazione. La maggior parte delle mutazioni non apporta alcuna differenza, o portare a proteine non funzionali, ma alcune rare mutazioni possono dare origine a una proteina più efficiente. Essenzialmente, sia le proteine funzionali che quelle non funzionali vengono prodotte durante l'evoluzione, ma secondo la teoria darwiniana della "sopravvivenza del più adatto", solo le proteine funzionali vengono mantenute e le proteine non funzionali alla fine si estinguono.
Che aspetto ha un codice "funzionale"? La risposta non è semplice. Infatti, il numero di codici di una proteina funzionale, anche una semplice proteina, è enorme, più grande della dimensione dell'universo. Però, utilizzando tecniche di analisi dei dati, è possibile ricercare pattern nascosti in tutti i codici funzionali per cercare alcune caratteristiche unificanti. Per esempio, il canale "floppy" nella proteina ha caratteristiche interessanti e peculiari, e una mutazione ad un'estremità del canale ha effetti a lungo raggio che possono influenzare fortemente il mantenimento delle mutazioni di altri amminoacidi distanti.
"Nel futuro, abbiamo in programma di esplorare come applicare questo studio a proteine reali, come chinasi, " ha affermato il capogruppo Tsvi Tlusty, un corrispondente nello studio. "Inoltre, lo studio apre strade per studiare l'evoluzione di altre funzioni proteiche, come il riconoscimento molecolare. Utilizzando enormi database, che sono stati sviluppati in anni di ricerca, può probabilmente scoprire alcuni fenomeni sottostanti sull'evoluzione delle proteine."