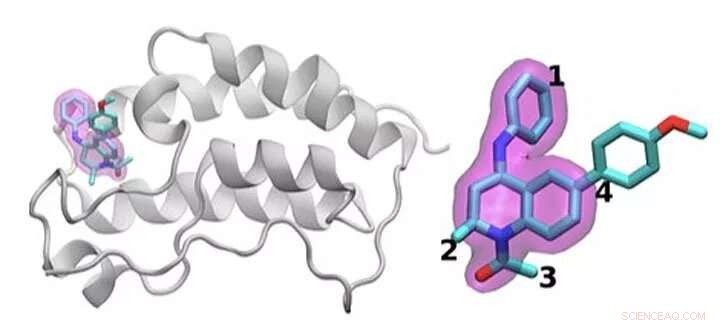
Uno schema della proteina BRD4 legata a uno dei 16 farmaci basati sullo stesso scaffold di tetraidrochinolina (evidenziato in magenta). Le regioni che sono chimicamente modificate tra i farmaci studiati in questo studio sono etichettate da 1 a 4. Tipicamente, viene apportata solo una piccola modifica alla struttura chimica da un farmaco all'altro. Questo approccio conservativo consente ai ricercatori di esplorare perché un farmaco è efficace mentre un altro non lo è. Credito:Brookhaven National Laboratory
Identificare il trattamento farmacologico ottimale è come colpire un bersaglio mobile. Per fermare la malattia, i farmaci a piccole molecole si legano strettamente a una proteina importante, bloccando i suoi effetti nel corpo. Anche i farmaci approvati di solito non funzionano in tutti i pazienti. E nel tempo, agenti infettivi o cellule cancerose possono mutare, rendendo inutile un farmaco una volta efficace.
Un problema fisico fondamentale è alla base di tutti questi problemi:ottimizzare l'interazione tra la molecola del farmaco e il suo bersaglio proteico. Le variazioni nelle molecole candidate ai farmaci, la gamma di mutazioni nelle proteine e la complessità complessiva di queste interazioni fisiche rendono difficile questo lavoro.
Shantenu Jha del Brookhaven National Laboratory del Dipartimento dell'Energia (DOE) e della Rutgers University guida un team che cerca di semplificare i metodi di calcolo in modo che i supercomputer possano sostenere parte di questo immenso carico di lavoro. Hanno trovato una nuova strategia per affrontare una parte:differenziare il modo in cui i farmaci candidati interagiscono e si legano con una proteina mirata.
Per il loro lavoro, Jha e i suoi colleghi hanno vinto lo scorso anno il premio IEEE International Scalable Computing Challenge (SCALE), che riconosce soluzioni di calcolo scalabili a problemi scientifici e ingegneristici del mondo reale.
Per progettare un nuovo farmaco, un'azienda farmaceutica potrebbe iniziare con una libreria di milioni di molecole candidate che si restringono alle migliaia che mostrano un legame iniziale con una proteina bersaglio. Affinare queste opzioni a un farmaco utile che può essere testato sull'uomo può comportare esperimenti approfonditi per aggiungere o sottrarre gruppi di atomi in punti chiave della molecola e testare come ciascuno di questi cambiamenti altera il modo in cui la piccola molecola e la proteina interagiscono.
Le simulazioni possono aiutare in questo processo. Più grandi, supercomputer più veloci e algoritmi sempre più sofisticati possono incorporare fisica realistica e calcolare le energie di legame tra varie piccole molecole e proteine. Tali metodi possono consumare notevoli risorse computazionali, però, per ottenere la necessaria precisione. Anche le simulazioni utili per il settore devono fornire risposte rapide. A causa del tiro alla fune tra precisione e velocità, i ricercatori innovano costantemente, sviluppando algoritmi più efficienti e migliorando le prestazioni, dice Jha.
Questo problema richiede anche la gestione delle risorse computazionali in modo diverso rispetto a molti altri problemi su larga scala. Invece di progettare una singola simulazione scalabile per utilizzare un intero supercomputer, i ricercatori eseguono simultaneamente molti modelli più piccoli che si modellano a vicenda e la traiettoria dei calcoli futuri, una strategia nota come calcolo basato sull'insieme, o flussi di lavoro complessi.
"Pensa a questo come al tentativo di esplorare un paesaggio aperto molto ampio per cercare di trovare dove potresti essere in grado di ottenere il miglior candidato alla droga, " dice Jha. In passato, i ricercatori hanno chiesto ai computer di navigare in questo panorama facendo scelte statistiche casuali. Ad un punto di decisione, metà dei calcoli potrebbe seguire un percorso, l'altra metà un'altra.
Jha e il suo team cercano invece modi per aiutare queste simulazioni a imparare dal paesaggio. L'acquisizione e la condivisione dei dati in tempo reale non è facile, Jha dice, "ed è quello che ha richiesto una parte dell'innovazione tecnologica per fare su larga scala". Lui e il suo team di Rutgers stanno collaborando con il gruppo di Peter Coveney presso l'University College di Londra su questo lavoro.
Per testare questa idea, hanno utilizzato algoritmi che prevedono l'affinità di legame e hanno introdotto versioni semplificate in un framework HTBAC, per il calcolatore dell'affinità di legame ad alta produttività. Una tale calcolatrice, noto come ESMACS, li aiuta a eliminare le molecole che si legano male a una proteina bersaglio. L'altro, CRAVATTE, è più accurato ma più limitato nell'ambito e richiede 2,5 volte più risorse di calcolo. Ciò nonostante, può aiutare i ricercatori a ottimizzare una promettente interazione tra un farmaco e una proteina. Il framework HTBAC li aiuta a implementare questi algoritmi in modo efficiente, salvare l'algoritmo più intensivo per le situazioni in cui è necessario.
Il team ha dimostrato l'idea esaminando 16 farmaci candidati da una libreria di molecole presso GlaxoSmithKline (GSK) con il loro obiettivo, BRD4-BD1:una proteina importante nel cancro al seno e nelle malattie infiammatorie. I farmaci candidati avevano la stessa struttura centrale ma differivano in quattro aree distinte attorno ai bordi della molecola.
In questo studio iniziale il team ha eseguito migliaia di processi contemporaneamente su 32, 000 core su Blue Waters, un supercomputer della National Science Foundation (NSF) presso l'Università dell'Illinois a Urbana-Champaign. Hanno eseguito calcoli simili su Titano, il supercomputer Cray XK7 presso l'Oak Ridge Leadership Computing Facility, una struttura per gli utenti dell'Office of Science del DOE. Il team ha distinto con successo il legame di questi 16 farmaci candidati, la più grande simulazione di questo tipo fino ad oggi. "Non abbiamo solo raggiunto una scala senza precedenti, " dice Jha. "Il nostro approccio mostra la capacità di differenziare".
Hanno vinto il premio SCALE per questa prova iniziale di concetto. La sfida ora, Jha dice, si sta assicurando che funzioni non solo per BRD4 ma anche per altre combinazioni di molecole di farmaci e bersagli proteici.
Se i ricercatori possono continuare ad espandere il loro approccio, tali tecniche potrebbero eventualmente aiutare ad accelerare la scoperta di farmaci e consentire una medicina personalizzata. Ma per esaminare problemi più realistici, avranno bisogno di più potenza di calcolo. "Siamo nel mezzo di questa tensione tra uno spazio chimico molto grande che noi, in linea di principio, bisogno di esplorare, e, risorse del computer purtroppo limitate." dice Jha.
Anche se il supercalcolo si espande verso l'esascala, gli scienziati computazionali possono più che colmare il divario aggiungendo una fisica più realistica ai loro modelli. Per il prossimo futuro, i ricercatori dovranno essere pieni di risorse per aumentare questi calcoli. La necessità è la madre dell'innovazione, Jha dice, proprio perché la scienza molecolare non avrà la quantità ideale di risorse computazionali per effettuare simulazioni.
Ma l'exascale computing può aiutarli ad avvicinarsi ai loro obiettivi. Oltre a lavorare con University College London e GSK, Jha e i suoi colleghi stanno collaborando con Rick Stevens dell'Argonne National Laboratory e il team CANcer Distributed Learning Environment (CANDLE). Questo progetto di co-design all'interno dell'Exascale Computing Project del DOE sta costruendo reti neurali profonde e tecniche generali di apprendimento automatico per studiare il cancro. Gli algoritmi e il software all'interno di HTBAC potrebbero integrare l'attenzione di CANDLE su questi approcci.
Questa più ampia collaborazione tra il gruppo di Jha, il team CANDLE e il laboratorio di John Chodera presso il Memorial Sloan-Kettering Cancer Center hanno portato al progetto INSPIRE (Integrated and Scalable Prediction of Resistance). Questo team ha già eseguito simulazioni sul supercomputer Summit del DOE presso l'Oak Ridge National Laboratory. Presto continuerà questo lavoro su Frontera, la macchina di comando della NSF presso l'Università del Texas presso il Texas Advanced Computing Center di Austin.
"Siamo affamati di maggiori progressi e maggiori miglioramenti metodologici, " Dice Jha. "Ci piacerebbe vedere come questi approcci piuttosto complementari potrebbero funzionare in modo integrato verso questa grande visione".