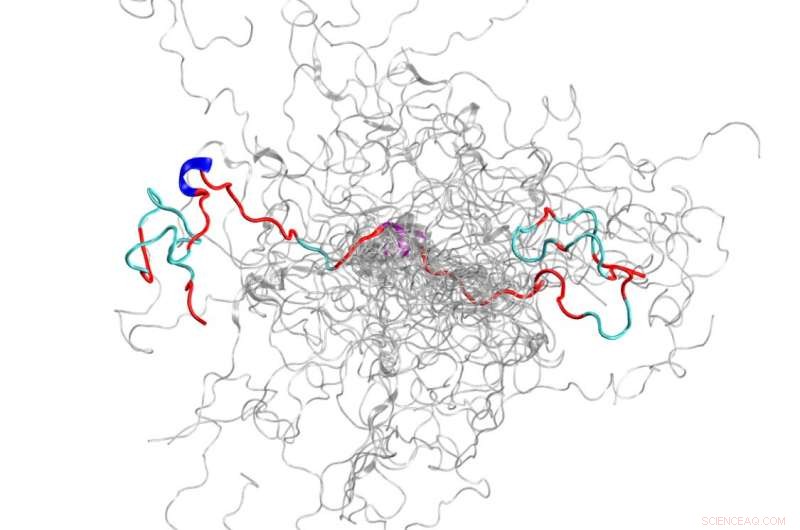
L'insieme configurazionale (una raccolta di strutture 3D) di una proteina intrinsecamente disordinata, l'N-terminale della chinasi c-Src, che è una delle principali proteine di segnalazione negli esseri umani. Credito:Oak Ridge National Laboratory, Dipartimento dell'Energia degli Stati Uniti
Utilizzando il supercomputer Titan e la Spallation Neutron Source presso l'Oak Ridge National Laboratory del Department of Energy, gli scienziati hanno creato il modello 3D più accurato mai realizzato di una proteina intrinsecamente disordinata, rivelando l'insieme delle sue strutture a livello atomico.
Come indica il nome, un IDP non adotta un ordinato, struttura statica come altre proteine; Invece, è flessibile e può adottare più strutture 3D. Questa mancanza di una struttura unica è necessaria per la funzione biologica dell'IDP, ma lo rende tecnicamente difficile da studiare. Gli IDP possono essere una proteina intera o un dominio di una proteina altrimenti strutturata, e costituiscono una gran parte dell'umano, microbo, e proteine vegetali.
Loukas Petridis, uno scienziato del personale presso il Centro di biofisica molecolare dell'ORNL, ha indirizzato un team di ricercatori verso un nuovo modo di creare modelli fisici accurati di tali biosistemi flessibili, che può portare a una migliore comprensione delle loro funzioni biologiche. Negli ultimi tre anni, il team ha combinato esperimenti di diffusione di neutroni con simulazioni di dinamica molecolare (MD) di campionamento potenziate così complesse dal punto di vista computazionale da richiedere la potenza di elaborazione di Titano, il Cray XK7 da 27 petaflop recentemente dismesso presso l'Oak Ridge Leadership Computing Facility, un DOE Office of Science User Facility presso ORNL.
"Studiare questi sfollati interni è abbastanza difficile, da entrambi i punti di vista degli esperimenti e della modellazione, " disse Utsab Shrestha, l'autore principale del documento del team, recentemente pubblicato su Atti dell'Accademia Nazionale delle Scienze . "Non ci abbiamo pensato solo da esperimenti o simulazioni da soli, abbiamo pianificato in modo da sinergizzare entrambi questi approcci, combinandoli in modo da poter ottenere informazioni più precise sugli sfollati interni. Nello specifico, le simulazioni ci hanno aiutato a generare un insieme accurato di IDP a risoluzione atomica, che è difficile da determinare dai soli esperimenti."
Tipicamente, i ricercatori conducono esperimenti come la diffusione di neutroni a piccolo angolo, diffusione di raggi X a piccolo angolo, o risonanza magnetica nucleare per sondare sistemi biologici flessibili. Però, questi metodi non forniscono un quadro dettagliato a livello atomico delle strutture 3-D di un IDP, noto come il suo insieme configurazionale. Per di più, possono produrre solo dati medi di insieme, piuttosto che le specifiche configurazioni della struttura proteica sottostante. Gli scienziati hanno anche eseguito simulazioni al computer di IDP e le hanno confrontate con tali esperimenti, sperando di ottenere gli stessi risultati al fine di verificare l'accuratezza dei loro modelli.
"Ma finiscono per non essere d'accordo con gli esperimenti, " disse Petridis. "E a causa della discrepanza tra le simulazioni e gli esperimenti, devono ripesare le simulazioni:devono regolare i risultati della simulazione per farli corrispondere agli esperimenti, il che è frustrante. Questo era lo stato dell'arte fino al nostro lavoro".
Le simulazioni al computer MD condotte da Shrestha hanno utilizzato metodi di campionamento avanzati che sono riusciti a far corrispondere non solo gli esperimenti di diffusione di neutroni, condotti da Viswanathan Gurumoorthy e dai suoi colleghi di SNS, un DOE Office of Science User Facility presso ORNL, ma anche dati NMR precedentemente pubblicati. Queste simulazioni MD utilizzano la fisica per determinare come si muovono le proteine. La chiave del successo del team è stata l'esecuzione di molte simulazioni MD in parallelo su Titano, consentendo alle simulazioni di comunicare tra loro e scambiarsi informazioni.
"Questo è molto importante perché consente alla simulazione di campionare uno spazio configurazionale più ampio, esplorare più strutture tridimensionali in modo più efficiente, " ha detto Petridis. "Ecco perché questo MD a campionamento avanzato può produrre risultati che la normale simulazione MD non può. Dovremmo eseguire una normale simulazione MD per anni per ottenere gli stessi risultati".
L'IDP che il team ha scelto di studiare è il dominio N-terminale della chinasi c-Src, che è una delle principali proteine di segnalazione negli esseri umani. Le mutazioni in questa complessa proteina sono state correlate al cancro, che lo rende anche un importante bersaglio farmacologico. Durante la mappatura di questo dominio precedentemente oscuro, gli scienziati sono stati in grado di scoprire nuove informazioni sulle sue strutture 3D che i metodi precedenti non avevano mostrato. Per esempio, sebbene sia in gran parte disordinato, questa proteina forma strutture ordinate transitorie, come le eliche.
"La combinazione di esperimenti di diffusione di neutroni e simulazione è molto potente, " ha detto Petridis. "La convalida delle simulazioni rispetto agli esperimenti di diffusione di neutroni è essenziale per avere fiducia nei risultati della simulazione. Le simulazioni convalidate possono quindi fornire informazioni dettagliate che non sono ottenute direttamente dagli esperimenti".
Il modello dettagliato al computer dell'insieme di strutture 3-D dell'IDP apre la porta a ulteriori sperimentazioni. Per esempio, gli scienziati potrebbero simulare l'effetto della fosforilazione (l'aggiunta di un gruppo fosfato alla proteina che può regolare la funzione della proteina) per vedere quali cambiamenti strutturali avvengono nella chinasi c-Src che potrebbero influenzare la sua funzione. Si potrebbe anche esaminare il ruolo delle mutazioni:se un ricercatore modifica un amminoacido nella catena, come influisce questo sulla struttura o sull'insieme delle strutture?
"Ci sono molte domande senza risposta per la c-Src chinasi in particolare a cui si potrebbe rispondere in termini di interazioni con altri partner:l'effetto della fosforilazione, l'effetto delle mutazioni, " disse Petridis.
Al di là dei potenziali usi scientifici per il modello stesso, Petridis vede l'opportunità di applicare l'uso del calcolo ad alte prestazioni per l'esecuzione di MD a campionamento avanzato per studiare le strutture di molti altri importanti IDP, che potrebbe dare un'idea della loro funzione. E più in generale, il team vuole sviluppare tecnologie di simulazione in grado di riprodurre profili di diffusione di neutroni a piccolo angolo di sistemi biologici ancora più complessi.
"Non vogliamo indagare solo sulle proteine disordinate:vogliamo avere sistemi molto più grandi che contengano domini ordinati e disordinati che potrebbero interagire con le membrane o il DNA, " disse Petridis. "La diffusione dei neutroni è, secondo me, la migliore tecnica sperimentale per sondare questi sistemi multicomponente, ad esempio, una proteina che interagisce con una membrana o una proteina che interagisce con il DNA. Ma, ancora, lo scattering di neutroni ha bisogno di simulazioni accurate per interpretare meglio i dati".