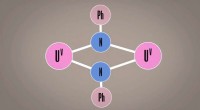
Strutture generate e reti di percorsi di reazione per le fasi di reazione previste per la reazione di Strecker (a) e reazione di Passerini (b). Le frecce bianche mostrano il percorso multifase corrispondente al meccanismo di reazione noto. Credito:Satoshi Maeda
I ricercatori hanno superato i limiti computazionali per prevedere i materiali di partenza delle reazioni multifase utilizzando solo informazioni sulla molecola del prodotto target. Il loro studio è pubblicato su JACS Au .
Hai mai catturato solo la fine di uno show televisivo e ti sei chiesto come è andata la storia fino a quel finale? Allo stesso modo, i chimici spesso hanno in mente una molecola desiderata e si chiedono che tipo di reazione potrebbe produrla. I ricercatori del Gruppo Maeda presso l'Institute for Chemical Reaction Design and Discovery (ICReDD) e l'Università di Hokkaido hanno sviluppato un metodo in grado di prevedere la "storia" (cioè i materiali di partenza e i percorsi di reazione) di reazioni chimiche multifase utilizzando solo informazioni su il "finale" (cioè le molecole del prodotto).
Prevedere la ricetta per una molecola di prodotto target, senza altra conoscenza che la molecola stessa, sarebbe un potente strumento per accelerare la scoperta di nuove reazioni. Il gruppo Maeda ha sviluppato in precedenza un metodo computazionale che è riuscito a prevedere le reazioni in un solo passaggio in questo modo. Tuttavia, l'espansione a reazioni multifase porta a un drammatico aumento del numero di possibili percorsi di reazione, ciò che è noto come esplosione combinatoria. Questo forte aumento della complessità si traduce in costi di calcolo proibitivi.
Per superare questa limitazione, i ricercatori hanno sviluppato un algoritmo che riduce il numero di percorsi che devono essere esplorati scartando i percorsi meno praticabili in ogni fase della reazione. Dopo aver calcolato tutti i percorsi possibili per un passo indietro nella reazione, un metodo di analisi cinetica valuta quanto bene ciascun percorso produce la molecola bersaglio. I percorsi di reazione che non producono la molecola bersaglio al di sopra di una soglia percentuale prestabilita sono ritenuti non sufficientemente significativi e non vengono ulteriormente esplorati.
Questo ciclo di esplorazione, valutazione e scarto dei percorsi di reazione viene ripetuto per ogni passo indietro in una reazione a più stadi e mitiga l'esplosione combinatoria che si verificherebbe normalmente, rendendo più fattibili il calcolo delle reazioni a più stadi. I metodi precedenti erano limitati a reazioni a fase singola, mentre questo nuovo metodo era in grado di prevedere reazioni che coinvolgevano più di sei fasi, segnando un notevole salto di capacità.
Come test di verifica del concetto, i ricercatori hanno testato il metodo su due note reazioni multifase, le reazioni di Strecker e Passerini. Migliaia di candidati materiali di partenza sono stati proposti per ciascuna reazione, che sono stati filtrati ai candidati più promettenti in base alla stabilità e alla resa del prodotto. Fondamentalmente, tra i candidati proposti c'erano i materiali di partenza ben noti per ciascuna reazione, a conferma della capacità della tecnica di identificare materiali di partenza sperimentalmente validi solo dalla molecola del prodotto target.
Sebbene siano necessari ulteriori lavori per consentire la previsione di sistemi ancora più grandi e complessi, i ricercatori prevedono che questa svolta nella gestione dei processi a più fasi accelererà la scoperta di nuove reazioni chimiche.
"Questo lavoro fornisce un approccio unico, poiché è la prima volta che è possibile eseguire previsioni inverse di reazioni multi-step utilizzando calcoli chimici quantistici senza utilizzare alcuna conoscenza o dati sulla reazione", ha affermato il professor Satoshi Maeda. "Ci aspettiamo che questa tecnica consentirà la scoperta di trasformazioni chimiche del tutto inimmaginabili, nel qual caso ci sono poche conoscenze o dati sperimentali da utilizzare". + Esplora ulteriormente