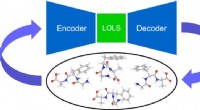
I ricercatori della Cleveland Clinic e dell'IBM hanno recentemente pubblicato i risultati sul Journal of Chemical Theory and Computation ciò potrebbe gettare le basi per l'applicazione di metodi di calcolo quantistico alla previsione della struttura delle proteine.
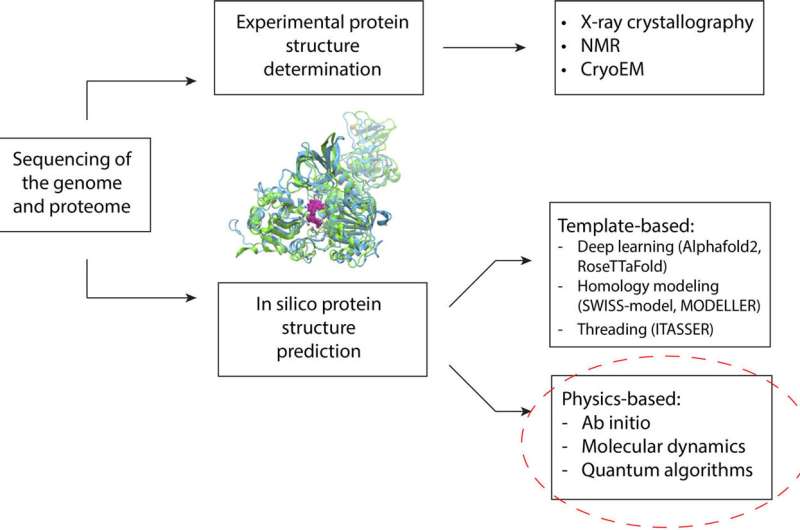
Per decenni, i ricercatori hanno sfruttato approcci computazionali per prevedere le strutture delle proteine. Una proteina si ripiega in una struttura che determina il modo in cui funziona e si lega ad altre molecole nel corpo. Queste strutture determinano molti aspetti della salute e delle malattie umane.
Prevedendo accuratamente la struttura di una proteina, i ricercatori possono comprendere meglio come si diffondono le malattie e quindi come sviluppare terapie efficaci. Bryan Raubenolt, ricercatore post-dottorato della Cleveland Clinic, Ph.D. e il ricercatore IBM Hakan Doga, Ph.D. ha guidato un team per scoprire come l'informatica quantistica può migliorare i metodi attuali.
Negli ultimi anni, le tecniche di apprendimento automatico hanno compiuto progressi significativi nella previsione della struttura delle proteine. Questi metodi fanno affidamento sui dati di addestramento (un database di strutture proteiche determinate sperimentalmente) per fare previsioni. Ciò significa che sono vincolati dal numero di proteine che è stato loro insegnato a riconoscere. Ciò può portare a livelli di precisione inferiori quando i programmi/algoritmi incontrano una proteina mutata o molto diversa da quelle su cui sono stati addestrati, il che è comune nelle malattie genetiche.
Il metodo alternativo consiste nel simulare la fisica del ripiegamento delle proteine. Le simulazioni consentono ai ricercatori di esaminare le varie forme possibili di una determinata proteina e di trovare quella più stabile. La forma più stabile è fondamentale per la progettazione di farmaci.
La sfida è che queste simulazioni sono quasi impossibili su un computer classico, oltre una certa dimensione proteica. In un certo senso, aumentare la dimensione della proteina bersaglio è paragonabile all'aumento delle dimensioni di un cubo di Rubik. Per una piccola proteina con 100 aminoacidi, un computer classico avrebbe bisogno di un tempo pari all'età dell'universo per cercare in modo esaustivo tutti i possibili risultati, afferma il dottor Raubenolt.
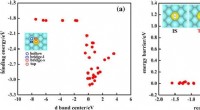
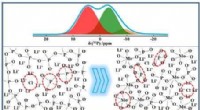
Per aiutare a superare queste limitazioni, il team di ricerca ha applicato un mix di metodi di calcolo quantistico e classico. Questo quadro potrebbe consentire agli algoritmi quantistici di affrontare le aree che rappresentano una sfida per l’informatica classica all’avanguardia, tra cui la dimensione delle proteine, il disordine intrinseco, le mutazioni e la fisica coinvolta nel ripiegamento delle proteine. Il quadro è stato convalidato prevedendo con precisione il ripiegamento di un piccolo frammento di una proteina del virus Zika su un computer quantistico, rispetto ai metodi classici all'avanguardia.
I risultati iniziali del quadro ibrido quantistico-classico hanno sovraperformato sia un metodo classico basato sulla fisica che AlphaFold2. Sebbene quest'ultimo sia progettato per funzionare al meglio con proteine più grandi, dimostra comunque la capacità di questo framework di creare modelli accurati senza fare affidamento direttamente su dati di addestramento sostanziali.
I ricercatori hanno utilizzato un algoritmo quantistico per modellare innanzitutto la conformazione a energia più bassa per la spina dorsale del frammento, che in genere è la fase più impegnativa dal punto di vista computazionale del calcolo. Sono stati quindi utilizzati approcci classici per convertire i risultati ottenuti dal computer quantistico, ricostruire la proteina con le sue catene laterali ed eseguire il perfezionamento finale della struttura con i campi di forza della meccanica molecolare classica.
Il progetto mostra uno dei modi in cui è possibile scomporre i problemi in parti, con metodi di calcolo quantistico che affrontano alcune parti e il calcolo classico altre, per una maggiore precisione.
"Una delle cose più singolari di questo progetto è il numero di discipline coinvolte", afferma il dottor Raubenolt. "Le competenze del nostro team spaziano dalla biologia e chimica computazionale, alla biologia strutturale, all'ingegneria del software e dell'automazione, alla fisica atomica e nucleare sperimentale, alla matematica e, naturalmente, all'informatica quantistica e alla progettazione di algoritmi. Sono state necessarie le conoscenze di ciascuna di queste aree per creare un quadro computazionale in grado di imitare uno dei processi più importanti per la vita umana."
La combinazione di metodi di calcolo classici e quantistici da parte del team rappresenta un passo essenziale per far avanzare la nostra comprensione delle strutture proteiche e del modo in cui influiscono sulla nostra capacità di curare e prevenire le malattie. Il team prevede di continuare a sviluppare e ottimizzare algoritmi quantistici in grado di prevedere la struttura di proteine più grandi e sofisticate.
"Questo lavoro rappresenta un importante passo avanti nell'esplorazione dei punti in cui le capacità del calcolo quantistico potrebbero mostrare punti di forza nella previsione della struttura delle proteine", afferma il dott. Doga. "Il nostro obiettivo è progettare algoritmi quantistici in grado di scoprire come prevedere le strutture delle proteine nel modo più realistico possibile."
Ulteriori informazioni: Hakan Doga et al, Una prospettiva sulla previsione della struttura delle proteine utilizzando i computer quantistici, Journal of Chemical Theory and Computation (2024). DOI:10.1021/acs.jctc.4c00067
Fornito da Cleveland Clinic