In un articolo recentemente pubblicato su Nature Communications , il gruppo di ricerca sulla modellizzazione delle proteine HUN-REN-ELTE (Istituto di chimica) ha gettato le basi per un metodo matematico che consente il confronto computerizzato delle strutture tridimensionali delle proteine. Il metodo è unico in quanto mentre le alternative disponibili finora prendevano in considerazione solo la posizione degli atomi, la nuova tecnica, chiamata LoCoHD (Local Composition Hellinger Distance), include anche le informazioni chimiche degli atomi.
Le proteine sono macchine molecolari che eseguono processi necessari al funzionamento delle cellule, agendo come interruttori molecolari, trascrivendo informazioni dal DNA, trasportando molecole piccole e grandi e regolando le reazioni chimiche legate al metabolismo. Ma affinché tutto ciò riesca, è necessario che la proteina in questione abbia la giusta conformazione spaziale, cioè una propria e corretta disposizione 3D.
Sono disponibili diversi metodi sperimentali (cristallografia a raggi X, spettroscopia di risonanza magnetica nucleare, microscopia crioelettronica) per determinare la disposizione degli atomi in una proteina e negli ultimi decenni i ricercatori sulle proteine hanno scoperto la forma di quasi 220.000 proteine. Questi risultati richiedono sempre più lo sviluppo di metodi computazionali in grado di analizzare queste disposizioni.
Uno di questi metodi è l'algoritmo chiamato LoCoHD, sviluppato da Zsolt Fazekas, un Ph.D. candidato alla Scuola di Chimica ELTE Hevesy György e ricercatore nel gruppo di ricerca del Dr. András Perczel. L'algoritmo confronta gli ambienti locali attorno agli amminoacidi nelle proteine in base alla loro natura chimica (ad esempio composizione elementare, carica, idrofobicità, ecc.).
Il metodo decide su una semplice scala da 0 a 1 quanto diverse sono le strutture in questione l'una dall'altra. Valori prossimi a 0 suggeriscono un'elevata somiglianza tra la disposizione atomica e le proprietà chimiche, mentre valori prossimi a 1 indicano che le proteine confrontate possono avere proprietà molto diverse. Il valore numerico risultante (una cosiddetta metrica) può quindi essere utilizzato per ottenere nuove informazioni sul sistema in esame.
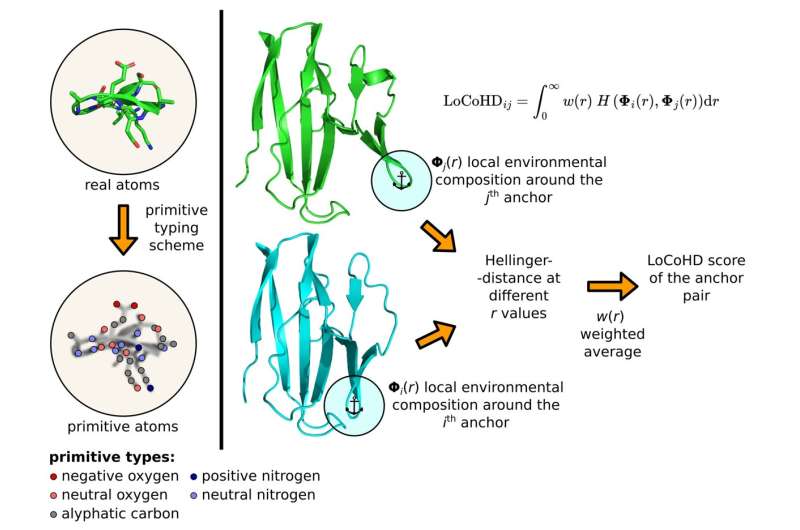
L'algoritmo utilizza un protocollo a più fasi per generare il numero che rappresenta le differenze strutturali. Nella prima fase converte gli atomi reali della proteina nei cosiddetti atomi primitivi. Questi possono essere rappresentati come posizioni virtualmente etichettate le cui etichette raccontano la natura chimica dell'atomo originale.
Così, ad esempio, un atomo primitivo può essere un "azoto carico positivamente", un "ossigeno carico negativamente", un "ossigeno carico neutro", un "carbonio aromatico", ecc. Le etichette vengono generate secondo un cosiddetto atomo primitivo schema di tipizzazione, che ci dice in modo tabellare come convertire gli atomi reali in atomi primitivi. L'utente può specificare liberamente questa tabella, fissando la risoluzione chimica del metodo.
Il secondo passo consiste nel determinare i punti di riferimento del confronto selezionando un sottoinsieme di atomi primitivi. Questi atomi primitivi speciali selezionati sono chiamati atomi di ancoraggio. Per ciascuna coppia di atomi di ancoraggio selezionata, l'algoritmo esegue un passaggio di confronto, il cui risultato fornisce la misura di dissomiglianza desiderata. Questi numeri possono essere utilizzati a livello locale oppure possono essere calcolati in media in un singolo descrittore che caratterizza l'intera proteina.
Nello studio, i ricercatori hanno sottolineato che il metodo può essere utilizzato anche nelle competizioni biennali CASP (Critical Assessment of Protein Structure Prediction), una competizione ben nota nel campo della ricerca sulle proteine. Durante questo evento, i concorrenti utilizzano diversi algoritmi per modellare la forma delle proteine con strutture non ancora pubblicate. I giudici CASP utilizzano una serie di metodi di confronto strutturale per valutare i contendenti, ma nessuno di questi tiene conto della chimica degli ambienti aminoacidici locali.
Utilizzando i dati del concorso CASP14 del 2020, i ricercatori hanno ora eseguito un’analisi comparativa di diverse proteine modellate, comprese le strutture previste dal metodo AlphaFold2 basato sull’intelligenza artificiale. Tra questi, hanno evidenziato l’analisi di una proteina del virus SARS-CoV-2 chiamata ORF8. Nelle strutture modellate di questa proteina, sono stati identificati ambienti di amminoacidi che differiscono significativamente nei loro modelli di interazione dagli ambienti trovati nella struttura sperimentale.
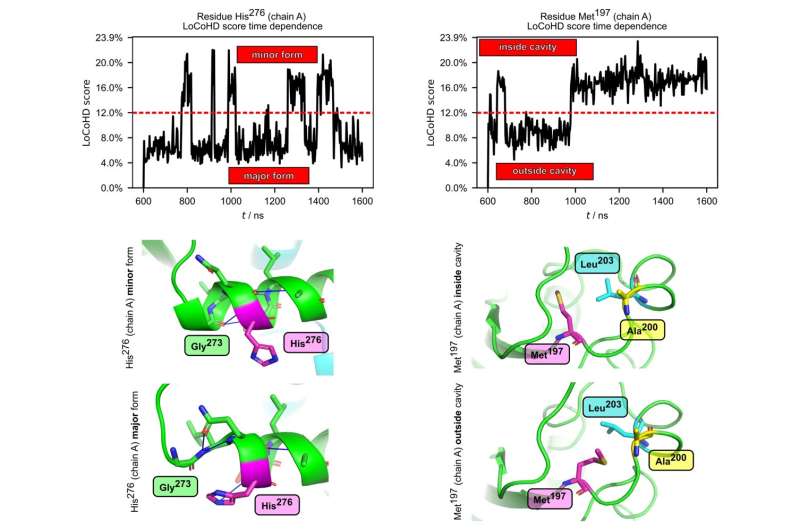
Oltre a studiare le strutture statiche, i ricercatori hanno anche testato se il metodo è adatto per analizzare il movimento interno delle proteine. Hanno utilizzato simulazioni in grado di riprodurre movimenti molecolari e dati estratti da insiemi strutturali. Uno dei sistemi oggetto di studio era la proteina podocina, che svolge funzioni vitali nel rene e le cui mutazioni possono causare condizioni gravi, spesso fatali.
Il metodo LoCoHD è stato utilizzato per identificare gli amminoacidi presenti nella proteina che subiscono importanti cambiamenti chimico-ambientali durante il movimento della podocina, che possono influenzarne sia la struttura che la funzione. Allo stesso modo, il metodo LoCoHD è stato applicato con successo nello studio della proteina capside dell'HIV-1, in cui è stato identificato un amminoacido fondamentale per la formazione dell'involucro virale.
Questi risultati non sono solo curiosità della ricerca, ma studiando le strutture proteiche in modo più efficace, possiamo avvicinarci a una migliore comprensione degli agenti patogeni che causano malattie gravi e allo sviluppo di farmaci e terapie efficaci.
Ulteriori informazioni: Zsolt Fazekas et al, LoCoHD:una metrica per confrontare gli ambienti locali delle proteine, Nature Communications (2024). DOI:10.1038/s41467-024-48225-0
Informazioni sul giornale: Comunicazioni sulla natura
Fornito dall'Università Eötvös Loránd