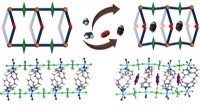
Gli ingegneri biomedici della Duke University hanno sviluppato una piattaforma di intelligenza artificiale che confronta autonomamente le molecole e impara dalle loro variazioni per anticipare le differenze di proprietà fondamentali per la scoperta di nuovi prodotti farmaceutici. La piattaforma fornisce ai ricercatori uno strumento più accurato ed efficiente per aiutare a progettare terapie e altre sostanze chimiche con proprietà utili.
La ricerca è stata pubblicata il 27 ottobre sul Journal of Cheminformatics .
Gli algoritmi di apprendimento automatico sono sempre più utilizzati per studiare e prevedere le proprietà biologiche, chimiche e fisiche di piccole molecole utilizzate nello sviluppo di farmaci e in altre attività di progettazione dei materiali. Questi strumenti possono aiutare i ricercatori a comprendere le principali proprietà "ADMET" di una molecola:come viene assorbita, distribuita, metabolizzata, escreta e la sua tossicità all'interno del corpo. Comprendendo queste diverse proprietà, i ricercatori possono identificare molecole per sviluppare nuove terapie più sicure ed efficaci.
Sebbene le piattaforme di apprendimento automatico esistenti consentano ai ricercatori di selezionare un numero molto maggiore di molecole rispetto a quanto sarebbe possibile realizzandole fisicamente tutte in laboratorio, essi possono solo prevedere le proprietà di una molecola alla volta, limitando la loro efficienza complessiva quando hanno il compito di identificare la molecola composto più ottimale.
Sebbene esistano alcuni altri approcci computazionali per eliminare questo passaggio aggiuntivo e confrontare direttamente le molecole, hanno una portata limitata. Ad esempio, metodi come la perturbazione dell’energia libera sono molto accurati ma così complessi dal punto di vista computazionale che possono valutare solo una manciata di molecole alla volta. Approcci come le coppie molecolari abbinate, d'altra parte, sono molto più veloci ma possono confrontare solo molecole molto simili, limitandone un uso più ampio.
Per affrontare questo problema, Reker e Zachary Fralish, un Ph.D. studente del laboratorio Reker, ha sviluppato DeepDelta, un approccio di apprendimento profondo in grado di confrontare in modo efficiente due molecole contemporaneamente e prevedere le differenze di proprietà tra loro anche se sono molto diverse.
"Facendo in modo che la rete impari da un confronto uno a uno, le dai più dati che se imparasse da una molecola alla volta", ha affermato Reker. "La piattaforma sta imparando a conoscere la struttura e le proprietà di ciascuna molecola individualmente, ma sta anche apprendendo le differenze tra le due e il modo in cui tali differenze influenzano le proprietà della molecola."
Il team ha testato la piattaforma DeepDelta confrontandola con due modelli all'avanguardia sul campo:Random Forest, un modello classico di apprendimento automatico ampiamente utilizzato, e ChemProp, una rete neurale profonda su cui si basa DeepDelta. Ciascun sistema ha confrontato due strutture molecolari conosciute e ha previsto 10 diverse proprietà ADMET, incluso il modo in cui le molecole vengono eliminate dai reni, le rispettive emivite e quanto bene possono essere metabolizzate dal fegato.
DeepDelta si è dimostrato significativamente più efficace e preciso nel prevedere e quantificare le differenze nelle proprietà molecolari tra le molecole rispetto alle piattaforme esistenti.
"La formazione sulle differenze molecolari consente a questo metodo di essere più accurato nel decidere se una nuova sostanza chimica è migliore o peggiore di quella attuale", ha affermato Fralish. "È come fare dei compiti che somigliano più a un test. Abbiamo anche ampliato notevolmente le dimensioni dei nostri set di dati mediante l'accoppiamento, essenzialmente dando ai nostri modelli più compiti, il che aiuta davvero le reti neurali affamate di dati a imparare di più."
Il team non vede l'ora di incorporare questo modello nel proprio lavoro mentre progetta potenziali nuove terapie e ottimizza i candidati farmaci esistenti.
"Con questo strumento, potremmo esaminare un farmaco che ha quasi superato l'approvazione della FDA, ma forse aveva problemi di tossicità epatica, quindi non ce l'ha fatta", ha detto Fralish. "DeepDelta potrebbe aiutare a identificare molecole che hanno quelle stesse buone proprietà ma nessuna tossicità epatica. Questo strumento apre molte opportunità aiutandoci a decidere quale sostanza chimica ha le migliori possibilità di fare ciò che vogliamo nel mondo reale, risparmiando tempo e denaro. "
Ulteriori informazioni: Zachary Fralish et al, DeepDelta:predire i miglioramenti ADMET dei derivati molecolari con il deep learning, Journal of Cheminformatics (2023). DOI:10.1186/s13321-023-00769-x
Fornito dalla Duke University