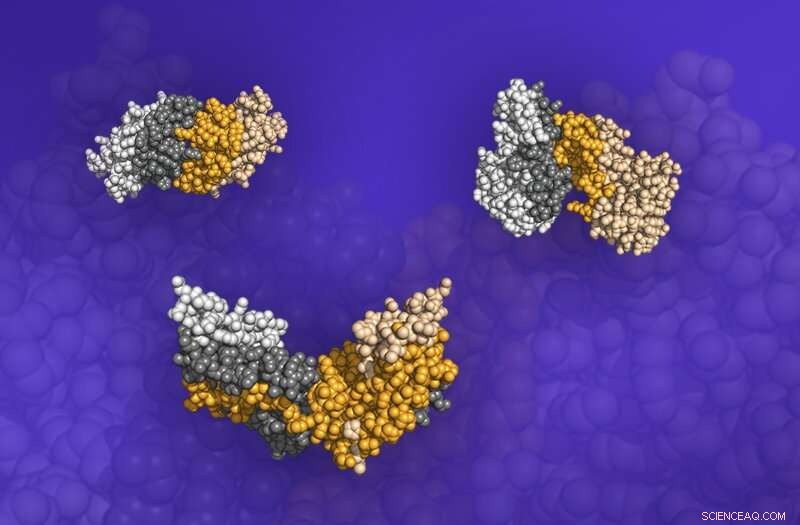
Tre dimeri, strutture proteiche costituite da due proteine legate, dal database Dockground. Le interfacce in cui le proteine si incontrano sono mostrate come regioni oscurate. Attestazione:ORNL
Può qualcosa di semplice come la forma determinare completamente se le proteine si legheranno o meno? Gli scienziati stanno commissionando dei supercomputer per scoprirlo.
Un team guidato da Sharon Glotzer, illustre professore e presidente del dipartimento di ingegneria chimica presso l'Università del Michigan (UM), ha utilizzato il supercomputer Summit da 200 petaflop presso l'Oak Ridge National Laboratory (ORNL) del Dipartimento dell'Energia degli Stati Uniti (DOE) per modellare le interazioni di blocco e chiave tra le proteine per studiarne i comportamenti di legame. I risultati, pubblicato in Materia morbida, ha rivelato che alcune proteine lo fanno, infatti, legare basandosi solo sulla forma.
"Abbiamo dimostrato che qualcosa di semplice come la forma è in grado di prevedere interazioni proteiche che a volte sono davvero complesse, "ha detto Jens Glaser, scienziato computazionale nel gruppo Advanced Computing for Chemistry and Materials presso l'Oak Ridge Leadership Computing Facility (OLCF). "Questa prima dimostrazione ci ha portato a credere che la forma sia stata un ingrediente non apprezzato in molti processi di assemblaggio delle proteine".
I risultati potrebbero avere numerose applicazioni nella ricerca biologica. Per esempio, l'approccio potrebbe essere utilizzato per esaminare i farmaci per le malattie o fornire agli scienziati informazioni su come utilizzare le proteine come elementi costitutivi per progettare nuovi materiali biologici.
"Questo studio entusiasmante dimostra il potere della complementarietà della forma nella previsione delle interfacce proteina-proteina, " ha detto la dottoressa Stephanie McElhinny, responsabile del programma presso il laboratorio di ricerca dell'esercito del comando di sviluppo delle capacità di combattimento dell'esercito degli Stati Uniti, riferendosi alla favorevole relazione spaziale tra due proteine di forma compatibile. "I modelli computazionali che prevedono con precisione queste interfacce supporteranno la progettazione futura di materiali avanzati a base di proteine con proprietà attive e reattive, come le plastiche a base di proteine che raccolgono la luce che potrebbero funzionare come una foglia artificiale per la generazione di energia".
I supercomputer rivelano che la forma è la chiave di alcune proteine
Affinché le proteine si leghino tra loro con successo, uno di loro funge da ligando, una molecola che si lega a una proteina bersaglio, e uno di loro funge da recettore, la molecola che riceve il ligando. Questo processo comporta interazioni chimiche complesse, in cui le molecole condividono legami e cambiano le loro configurazioni al momento del legame.
Il team di Glotzer voleva vedere se potevano prevedere questo legame molecolare basato solo sulla forma, ignorando le interazioni tra le proteine. Da un database di oltre 6, 000 coppie proteiche, il team ha testato 46 coppie note per legarsi l'una all'altra e ha simulato il loro assemblaggio su Summit. Il team ha eseguito le simulazioni nell'ambito del programma INCITE (Innovative and Novel Computational Impact on Theory and Experiment).
Come più palline da tennis lanciate contro un singolo bersaglio, le simulazioni hanno modellato più ligandi lanciati contro un singolo, recettore bersaglio fisso. Delle 46 coppie testate, hanno trovato 6 paia che hanno funzionato bene:più del 50% delle volte sono state assemblate con successo basandosi esclusivamente sulle loro forme complementari.
"Abbiamo esaminato le interfacce in cui le proteine si legano insieme per vedere quanto fossero simili alle loro interfacce della vita reale, e poi abbiamo determinato il cutoff per vedere quante coppie erano buoni predittori delle interfacce reali, " disse Fengyi Gao, dottorato di ricerca candidato presso UM. "Abbiamo scoperto che il 13% di queste coppie proteiche potrebbe legarsi solo in base alla forma".
Il team ha quindi costruito un modello di apprendimento automatico in grado di determinare quali proteine sono in grado di assemblarsi esclusivamente in base alla forma. La combinazione del loro modello iniziale con tali strumenti di apprendimento automatico li aiuterà a capire quali informazioni sono necessarie per le coppie proteiche che non possono essere assemblate in base alla sola complementarietà della forma.
Esecuzione di proteine in parallelo
Per modellare più processi di legame reversibili di 46 coppie proteiche sotto diversi parametri, avevano bisogno di due giorni di tempo computazionale e più di 3, 000 GPU, una quantità che solo un supercomputer come il Summit dell'OLCF potrebbe fornire. L'OLCF è un DOE Office of Science User Facility presso l'ORNL.
Come parte del codice computazionale HOOMD-blue utilizzato per eseguire le simulazioni, Glaser, che in precedenza era un assistente ricercatore nel gruppo di Glotzer alla UM, sviluppato un algoritmo che simulava le proteine in presenza di molte piccole particelle. Ma Glaser ha trovato un modo per modellare solo il movimento delle proteine a cui il team era interessato, evitando inutili e costosi calcoli per le molecole di solvente che li circondano.
"Ho eseguito il codice in parallelo in modo che molti parametri diversi, iterazioni dello stesso sistema, e diverse proteine potrebbero essere distribuite tra le GPU, " Ha detto Glaser. "Questo ci ha permesso di utilizzare facilmente le capacità di calcolo parallelo di Summit".
Usando il vertice, il team ha catturato sei coppie di proteine che si legano in base solo alla complementarietà della forma, con uno di loro che raggiunge la rilegatura più del 94 percento delle volte.
"È stato abbastanza sorprendente per noi che un modello così semplificato potesse selezionare correttamente solo quella posa che assumono tra le molte centinaia o più pose che competono, " Ha detto Glaser. "Ci aspettavamo che sarebbe stato necessario molto di più per riprodurre la vera posa di legame per queste coppie proteiche".
I modelli possono aiutare nello screening dei farmaci
Il team prevede di studiare più proteine che possono anche legarsi in base alla forma o formare strutture di ordine ancora più elevato. L'attuale studio del team ha esplorato solo dimeri proteici, che consistono in due proteine legate insieme, ma il team vuole conoscere i limiti di come le forme proteiche possono evolversi per formare strutture proteiche gerarchiche.
"Prima di fare questo studio, In realtà non mi aspettavo che le proteine potessero formare dimeri basandosi solo sulla forma, " disse Fengyi Gao, dottorato di ricerca candidato presso UM. "Ma ora, abbiamo scoperto che funziona, e possiamo studiare strutture più complesse o anche combinare questo con altri approcci, come l'apprendimento automatico, per vedere di quali funzionalità abbiamo bisogno per abilitare l'associazione corretta."
Il team spera di poter eventualmente prevedere il legame delle interfacce proteina-proteina nei cluster proteici o nelle strutture di cristallizzazione delle proteine.
"Pensiamo di poter adattare questo approccio a qualcosa come lo screening dei farmaci in futuro, " ha detto Gao. "In aggiunta a questo, speriamo che questo modello basato sulla forma possa servire come base per lo studio dell'assemblaggio proteico in generale".