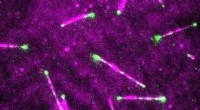
Utilizzando set di dati del metagenoma raccolti nel corso di diversi anni nei laghi d'acqua dolce del nord, un team guidato da ricercatori della Ohio State University e del Dipartimento dell'Energia degli Stati Uniti Joint Genome Institute (DOE JGI) ha scoperto 25 nuove sequenze di virofagi. Segnalato l'11 ottobre 2017 in Comunicazioni sulla natura , l'identificazione di queste nuove sequenze raddoppia efficacemente il numero di virofagi conosciuti dalla loro scoperta un decennio fa. Il team ha utilizzato i dati di una serie temporale metagenomica di 3 anni raccolti da Trout Bog Lake, una piccola palude acida nel Wisconsin, da collaboratori dell'Università del Wisconsin-Madison. Credito:Trina McMahon
Nei laghi d'acqua dolce, i microbi regolano il flusso di carbonio e determinano se i corpi idrici fungono da pozzi di carbonio o fonti di carbonio. Le alghe e i cianobatteri in particolare possono intrappolare e utilizzare il carbonio, ma la loro capacità di farlo può essere influenzata dai virus. I virus esistono in mezzo a tutti i batteri, di solito in un eccesso di 10 volte, e sono costituiti da varie dimensioni che vanno da virus giganti, a virus molto più piccoli noti come virofagi (che vivono in virus giganti e usano i loro macchinari per replicarsi e diffondersi). I virofagi possono cambiare il modo in cui un virus gigante interagisce con la sua cellula eucariotica ospite. Per esempio, se le alghe sono co-infettate da un virofago e da un virus gigante, il virofago limita la capacità del virus gigante di replicarsi in modo efficiente. Questo riduce l'impatto che un virus gigante ha sulla diversione dei nutrienti, permettendo alle alghe ospiti di moltiplicarsi, che potrebbe portare a fioriture algali più frequenti.
Utilizzando set di dati del metagenoma raccolti nel corso di diversi anni nei laghi d'acqua dolce del nord, un team guidato da ricercatori della Ohio State University e del Dipartimento dell'Energia degli Stati Uniti Joint Genome Institute (DOE JGI), una struttura per gli utenti dell'Office of Science del DOE, scoperto 25 nuove sequenze di virofagi. Segnalato l'11 ottobre 2017 in Comunicazioni sulla natura , l'identificazione di queste nuove sequenze raddoppia efficacemente il numero di virofagi conosciuti dalla loro scoperta un decennio fa.
"Di solito i set di dati del metagenoma sono una tantum, " ha detto lo scienziato DOE JGI e primo autore Simon Roux. "La gente aveva iniziato a vedere i virofagi nei metagenomi, ma nessuno ha avuto una lunga serie temporale fino ad ora. Era qui una volta? Sempre? Non lo abbiamo mai saputo veramente, ma è un'informazione fondamentale per capire la loro importanza".
Il lavoro è scaturito da una proposta del Community Science Program (CSP) riguardante i laghi d'acqua dolce del nord di KT (Trina) McMahon dell'Università del Wisconsin-Madison. Campioni di comunità microbiche nel lago Mendota e nel lago Trout Bog sono stati regolarmente raccolti per diversi anni nell'ambito del progetto North Temperate Lakes Long Term Ecological Research (NTL-LTER) della National Science Foundation, finanziato dall'NSF. Il sequenziamento e l'analisi di questi metagenomi dalle serie temporali di 3 e 5 anni consente ai ricercatori di identificare i membri della comunità, determinare le loro vie metaboliche, e seguire i cambiamenti nelle comunità nel corso di diversi anni.

Il team ha utilizzato i dati di una serie temporale di 5 anni raccolti dal Lago Mendota, un grande lago d'acqua dolce nel Wisconsin. Credito:McMahon Lab
Oltre a guardare le comunità microbiche, McMahon e Rex Malmstrom, capo del gruppo DOE JGI Micro-Scale Applications, ha chiesto al collaboratore Matt Sullivan della Ohio State University se fosse interessato a utilizzare gli stessi set di dati metagenomici per esaminare l'ecologia virale dei laghi. Roux ha iniziato a estrarre i set di dati mentre era ancora un borsista post-dottorato con il laboratorio Sullivan. "Sapevo che c'erano molti virus nei dati della sequenza, ma non che alcuni virus fossero essi stessi ospiti di altri virus, " ha detto Malmstrom. "Con i dati delle serie temporali potremmo fare di più che assemblare il genoma e costruire alberi filogenetici, i dati ci hanno permesso di esaminare la variazione genetica all'interno delle popolazioni e cercare modelli di co-occorrenza e abbondanza tra i virofagi e i loro ospiti giganti del virus. Con così tanti punti temporali nel set di dati, puoi trovare connessioni forti."
Trina McMahon, i cui set di dati CSP sono stati la base di questo lavoro, dice che avere le informazioni sull'ecologia virale aiuta a formare un quadro più completo dell'ecosistema. "Siamo entusiasti di avere un altro pezzo del puzzle. I virus stanno chiaramente giocando un ruolo importante nel plasmare la composizione della comunità e quindi il funzionamento, dell'intero ecosistema lacustre. Il mio laboratorio non ha le competenze per affrontare da solo i virus, quindi la collaborazione con Simon e Matt Sullivan è così importante. Il nostro obiettivo a lungo termine è imparare abbastanza sulle forze che controllano l'assemblea e le dinamiche della comunità, così come i tratti ecologici di ogni lignaggio, in order to create more predictive models about how freshwater lakes will respond to climate and land-use change, at an ecosystem scale."
Aside from doubling the number of virophages in public databases, the time series allowed Roux and his colleagues to see the viruses' ecological profiles - if factors such as the seasons or abundance of particular microbes influenced their own presence. Through co-occurrence analysis, the researchers associated the virophages with sequences of known lineages of giant viruses, and proposed the existence of 3 new groups of candidate giant viruses infected by virophages. These co-occurrence analyses also allowed them to find putative associations between the giant virus sequences and specific eukaryotic hosts.
"These findings are correlation-based, " noted Roux, "but it's a good example of a metagenomics use case. Metagenomes helped us not only discover new viral diversity and determine what it should do in the ecosystem, but it helps us design hypothesis and follow-up experiments about virus-host interactions so we're not just throwing out a wide net blindly."