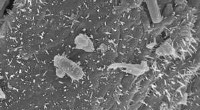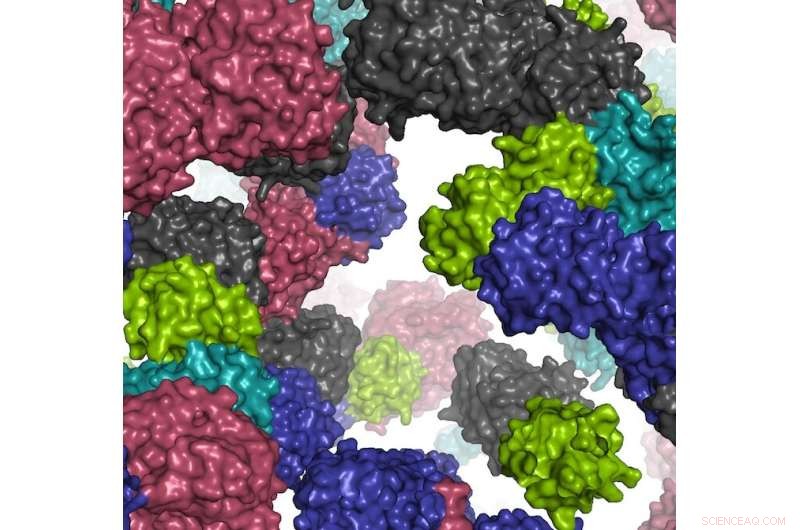
Un frammento dell'ambiente cellulare simulato. Credito:Ilya Vakser
Un rapporto fondamentale dell'Università del Kansas che apparirà questa settimana negli Proceedings of the National Academy of Sciences propone una nuova tecnica per modellare la vita molecolare con i computer.
Secondo l'autore principale Ilya Vakser, direttore del Programma di biologia computazionale e del Centro di biologia computazionale e professore di bioscienze molecolari presso la KU, l'indagine sulla modellazione al computer dei processi vitali è un passo importante verso la creazione di una simulazione funzionante di una cellula vivente a risoluzione atomica . Il progresso promette nuove intuizioni sulla biologia fondamentale di una cellula, nonché un trattamento più rapido e preciso delle malattie umane.
"È circa decine o centinaia di migliaia di volte più veloce delle tecniche di risoluzione atomica esistenti", ha detto Vakser. "Ciò offre opportunità senza precedenti per caratterizzare meccanismi fisiologici che ora sono ben oltre la portata della modellazione computazionale, per ottenere informazioni dettagliate sui meccanismi cellulari e utilizzare queste conoscenze per migliorare la nostra capacità di curare le malattie".
Finora, uno dei principali ostacoli alla modellazione delle cellule tramite computer è stato il modo in cui avvicinarsi alle proteine e alle loro interazioni che sono al centro dei processi cellulari. Ad oggi, le tecniche consolidate per la modellazione delle interazioni proteiche sono dipese dal "docking proteico" o dalla "simulazione molecolare".
Secondo i ricercatori, entrambi gli approcci presentano vantaggi e svantaggi. Sebbene gli algoritmi di ancoraggio proteico siano ottimi per campionare le coordinate spaziali, non tengono conto della "coordinata temporale" o della dinamica delle interazioni proteiche. Al contrario, le simulazioni molecolari modellano bene la dinamica, ma queste simulazioni sono troppo lente oa bassa risoluzione.
"Il nostro studio proof-of-concept unisce le due metodologie di modellazione, sviluppando un approccio che può raggiungere tempi di simulazione senza precedenti con risoluzione di tutti gli atomi", hanno scritto gli autori.
I collaboratori di Vakser sull'articolo erano Sergei Grudinin dell'Università di Grenoble Alpes in Francia; Eric Deeds dell'Università della California-Los Angeles; Nathan Jenkins, dottorando della KU, e Petras Kundrotas, assistente professore di ricerca presso il programma di biologia computazionale della KU.
Dopo aver concettualizzato il modo migliore per combinare i vantaggi dei due approcci di modellazione delle proteine, il team ha sviluppato e codificato un algoritmo per guidare la nuova simulazione.
"La sfida più difficile è stata sviluppare l'algoritmo che riflettesse adeguatamente la semplice idea di base dell'approccio", ha affermato Vakser.
Ma una volta raggiunta questa svolta, potrebbero iniziare a convalidare la nuova procedura.
"Il paradigma era facile, un colpo di chiarezza", ha detto Vakser.
"Gli approcci di simulazione esistenti trascorrono la maggior parte del tempo di calcolo viaggiando in aree del sistema a bassa probabilità o ad alta energia. Sappiamo tutti dove si trovano queste aree. Invece, l'idea era di campionare, o viaggiare, solo nelle aree ad alta -probabilità, aree a bassa energia, e saltare quelle a bassa probabilità stimando i tassi di transizione tra gli stati ad alta probabilità.Il paradigma è antico quanto la stessa modellazione biomolecolare ed è stato ampiamente utilizzato sin dagli albori dell'era della modellazione decenni fa."
Ma Vakser ha affermato che fino al nuovo articolo del suo team, l'approccio non era stato applicato alla cinetica delle interazioni proteiche nell'ambiente cellulare, oggetto del loro studio.
"Poiché ci sono molti meno stati ad alta probabilità rispetto a quelli a bassa probabilità, questo ci ha dato un enorme vantaggio nella velocità di calcolo, da decine a centinaia di migliaia di volte", ha detto Vakser. "Ciò è stato fatto senza un'apparente perdita di accuratezza. Si può sostenere che l'accuratezza sia stata ottenuta, perché il protocollo di simulazione si basa sulle tecniche di 'docking', progettate specificamente per caratterizzare gli assemblaggi proteici".
Il ricercatore della KU ha affermato che il suo metodo di simulazione cellulare potrebbe essere utilizzato per la ricerca sulla salute umana e il trattamento delle malattie con un nuovo livello di precisione.
"L'approccio può essere utilizzato per studiare i percorsi molecolari alla base dei meccanismi della malattia", ha affermato Vakser. "Può essere utilizzato per determinare gli effetti dannosi delle mutazioni genetiche dovute ai mutati modelli di associazioni proteiche:le mutazioni genetiche causano cambiamenti nella struttura delle proteine, che a loro volta influiscono sull'associazione delle proteine. Oppure potrebbe essere utilizzato per identificare gli obiettivi per la progettazione di farmaci mediante rilevare gli elementi critici nei modelli di associazione proteica."
Secondo Vakser, la nuova tecnica di simulazione offre molte promettenti vie di ricerca da esplorare in futuro.
"Tra questi ci sono l'adattamento dell'approccio alle interazioni proteiche con acidi nucleici, RNA e DNA", ha detto. "Inoltre, vorremmo tenere conto della flessibilità delle forme molecolari, correlare con lo spettro in rapido sviluppo degli studi sperimentali dell'ambiente cellulare e applicare la procedura a un modello di una cellula reale, con i suoi componenti molecolari effettivi impacchettati insieme". + Esplora ulteriormente