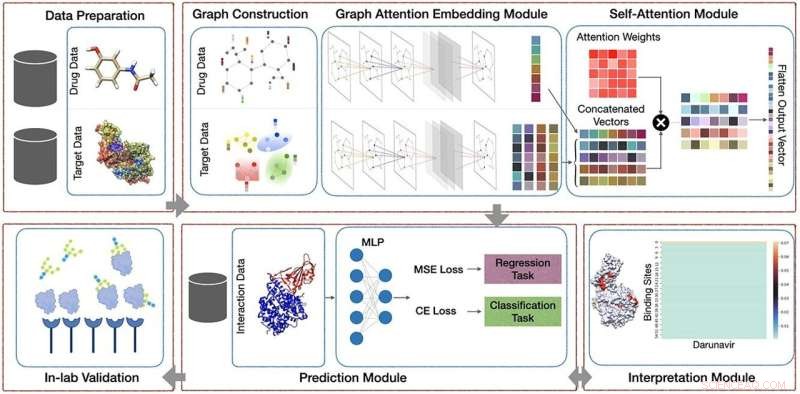
La nostra struttura proposta comprende cinque moduli principali:(1) modulo di preelaborazione che consiste nel trovare i siti di legame delle proteine; (2) Modulo di apprendimento profondo AttentionSiteDTI, in cui costruiamo rappresentazioni a grafo del SORRISO dei ligandi e dei siti di legame delle proteine, e creiamo una rete neurale convoluzionale del grafo armata di un meccanismo di pooling dell'attenzione per estrarre incorporamenti apprendibili dai grafi, nonché un auto- meccanismo di attenzione per apprendere la relazione tra ligandi e siti di legame delle proteine; (3) Modulo di previsione per prevedere l'interazione sconosciuta in una coppia farmaco-bersaglio, che può affrontare sia le attività di classificazione che di regressione; (4) Modulo di interpretazione per fornire una comprensione più approfondita di quali siti di legame di una proteina bersaglio hanno maggiori probabilità di legarsi con un dato ligando. (5) Convalide in laboratorio, dove confrontiamo i nostri risultati calcolati con le interazioni farmaco-bersaglio osservate sperimentalmente (misurate) in laboratorio per testare e validare il potenziale pratico del nostro modello proposto. Credito:Briefing in Bioinformatica (2022). DOI:10.1093/bib/bbac272
Lo sviluppo di medicinali salvavita può richiedere miliardi di dollari e decenni di tempo, ma i ricercatori dell'Università della Florida centrale mirano ad accelerare questo processo con un nuovo processo di screening dei farmaci basato sull'intelligenza artificiale che hanno sviluppato.
Utilizzando un metodo che modella le interazioni tra farmaci e proteine target utilizzando tecniche di elaborazione del linguaggio naturale, i ricercatori hanno ottenuto un'accuratezza fino al 97% nell'identificazione di promettenti farmaci candidati. I risultati sono stati pubblicati di recente sulla rivista Briefings in Bioinformatics .
La tecnica rappresenta le interazioni farmaco-proteina attraverso parole per ciascun sito di legame proteico e utilizza il deep learning per estrarre le caratteristiche che governano le complesse interazioni tra i due.
"Con l'aumento della disponibilità dell'IA, questo è diventato qualcosa che l'IA può affrontare", afferma il coautore dello studio Ozlem Garibay, un assistente professore presso il Dipartimento di ingegneria industriale e sistemi di gestione dell'UCF. "Puoi provare tante varianti di proteine e interazioni farmacologiche e scoprire quali hanno maggiori probabilità di legarsi o meno."
Il modello che hanno sviluppato, noto come AttentionSiteDTI, è il primo ad essere interpretabile utilizzando il linguaggio dei siti di legame proteico.
Il lavoro è importante perché aiuterà i progettisti di farmaci a identificare i siti critici di legame alle proteine insieme alle loro proprietà funzionali, che è la chiave per determinare se un farmaco sarà efficace.
I ricercatori hanno raggiunto il traguardo ideando un meccanismo di auto-attenzione che consente al modello di apprendere quali parti della proteina interagiscono con i composti del farmaco, ottenendo prestazioni predittive all'avanguardia.
La capacità di auto-attenzione del meccanismo funziona concentrandosi selettivamente sulle parti più rilevanti della proteina.
I ricercatori hanno convalidato il loro modello utilizzando esperimenti in laboratorio che hanno misurato le interazioni di legame tra composti e proteine e quindi hanno confrontato i risultati con quelli previsti dal loro modello dal punto di vista computazionale. Poiché i farmaci per il trattamento del COVID sono ancora interessanti, gli esperimenti includevano anche il test e la convalida di composti farmacologici che si legherebbero a una proteina spike del virus SARS-CoV2.
Garibay afferma che l'elevata concordanza tra i risultati di laboratorio e le previsioni computazionali illustra il potenziale di AttentionSiteDTI per lo screening preliminare di composti farmacologici potenzialmente efficaci e per accelerare l'esplorazione di nuovi farmaci e il riutilizzo di quelli esistenti.
"Questa ricerca ad alto impatto è stata possibile solo grazie alla collaborazione interdisciplinare tra ingegneria dei materiali e AI/ML e scienziati informatici per affrontare la scoperta relativa al COVID", afferma Sudipta Seal, coautore dello studio e presidente del Dipartimento di scienza e ingegneria dei materiali dell'UCF.
Mehdi Yazdani-Jahromi, uno studente di dottorato presso il College of Engineering and Computer Science dell'UCF e autore principale dello studio, afferma che il lavoro sta introducendo una nuova direzione nel pre-screening dei farmaci.
"Ciò consente ai ricercatori di utilizzare l'IA per identificare i farmaci in modo più accurato per rispondere rapidamente a nuove malattie", afferma Yazdani-Jahromi. "Questo metodo consente inoltre ai ricercatori di identificare il miglior sito di legame della proteina di un virus su cui concentrarsi nella progettazione di farmaci".
"Il prossimo passo della nostra ricerca sarà la progettazione di nuovi farmaci utilizzando la potenza dell'IA", afferma. "Questo naturalmente può essere il prossimo passo per essere preparati a una pandemia". + Esplora ulteriormente