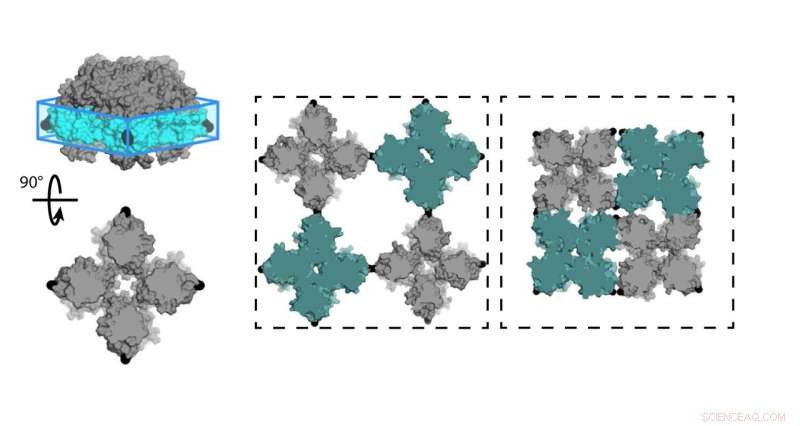
Chimici dell'Università della California, San Diego (UCSD) ha progettato un foglio di proteine (C98RhuA) che alterna diversi stati di porosità e densità. Le cellule del reticolo cristallino sono incernierate agli angoli del tetramero C98RhuA, permettendogli di girare e aprire o chiudere il poro. Credito:Robert Alberstein et al.
Ciò che fa fermare un proiettile al kevlar, a livello atomico?
Le proprietà dei materiali emergono dalla loro struttura molecolare o atomica, eppure molti dettagli tra il micro e il macro rimangono un mistero per la scienza. Gli scienziati stanno attivamente ricercando la progettazione razionale di architetture supramolecolari mirate, con l'obiettivo di ingegnerizzare le loro dinamiche strutturali e la loro risposta agli stimoli ambientali.
Un team di chimici dell'Università della California, San Diego (UCSD) ha ora progettato un cristallo proteico bidimensionale che alterna stati di porosità e densità variabili. Questa è la prima volta nella progettazione biomolecolare che ha combinato studi sperimentali con calcoli eseguiti su supercomputer. La ricerca, pubblicato ad aprile 2018 in Chimica della natura , potrebbe aiutare a creare nuovi materiali per l'energia rinnovabile, medicinale, purificazione dell'acqua, e altro ancora.
"Abbiamo fatto una vasta serie di simulazioni ed esperimenti di dinamica molecolare, che ha spiegato la base delle insolite dinamiche strutturali di queste proteine artificiali, in base alla quale siamo stati in grado di prendere decisioni razionali e alterare le dinamiche strutturali dell'assemblea, " ha detto il coautore dello studio Akif Tezcan, professore di chimica e biochimica alla UCSD.
Il team di Tezcan ha lavorato con la proteina L-ramnulosio-1-fosfato aldolasi (RhuA), che è stato modificato con amminoacidi cisteina nei suoi quattro angoli in posizione 98 (C98RhuA). Lui e il suo gruppo avevano precedentemente pubblicato un lavoro sull'autoassemblaggio di questo artificiale, architettura proteica bidimensionale, che ha detto ha mostrato un comportamento interessante chiamato auxeticity.
"Queste assemblee cristalline possono effettivamente aprirsi e chiudersi in modo coerente, "Tezcan ha detto. "Come fanno, si restringono o si espandono ugualmente nelle direzioni X e Y, che è l'opposto di ciò che fanno i materiali normali. Abbiamo voluto indagare a cosa sono dovuti questi moti e cosa li governa." Un esempio di auxeticità può essere visto nella Sfera di Hoberman, una palla giocattolo che si espande attraverso i suoi cardini a forbice quando separi le estremità.
"Il nostro obiettivo era quello di poter fare la stessa cosa, usando le proteine come elementi costitutivi, per creare nuove tipologie di materiali con proprietà avanzate, "Tezcan ha detto. "L'esempio che stiamo studiando qui era essenzialmente il frutto di quegli sforzi, dove abbiamo usato questa particolare proteina che ha una forma quadrata, che ci univamo l'un l'altro attraverso legami chimici che erano reversibili e fungevano da cardini. Ciò ha permesso a questi materiali di formare cristalli molto ben ordinati che erano anche dinamici grazie alla flessibilità di questi legami chimici, che ha finito per darci questi nuovi, proprietà emergenti."
Il controllo dell'apertura e della chiusura dei pori nei reticoli 2-D della proteina C98RhuA potrebbe catturare o rilasciare specifici bersagli molecolari utili per la somministrazione di farmaci o la creazione di batterie migliori con ulteriori ricerche, ha detto Tezcan. Oppure potrebbero passare selettivamente o bloccare il passaggio di molecole biologiche e filtrare l'acqua.

Il supercomputer Maverick è una risorsa dedicata di visualizzazione e analisi dei dati presso il Texas Advanced Computing Center, progettata con 132 unità di elaborazione grafica (GPU) NVIDIA Tesla K40 "Atlas" per la visualizzazione remota e il calcolo GPU per la comunità nazionale. Credito:TACC
"La nostra idea era quella di poter costruire materiali complessi, come ha fatto l'evoluzione, usando le proteine come elementi costitutivi, " ha detto Tezcan.
Il modo in cui lo ha fatto il team di Tezcan è stato quello di esprimere prima le proteine nelle cellule dei batteri di E. coli e purificarle, dopo di che hanno indotto la formazione dei legami chimici che effettivamente creano i cristalli di C98RhuA, che variano in funzione del loro stato di ossidazione, attraverso l'aggiunta di sostanze chimiche redox-attive.
"Una volta formati i cristalli, la grande caratterizzazione diventa l'apertura o la vicinanza dei cristalli stessi, " ha spiegato Tezcan, che è stato determinato attraverso l'analisi statistica di centinaia di immagini catturate mediante microscopia elettronica.
Gli esperimenti hanno lavorato di pari passo con il calcolo, principalmente simulazioni di tutti gli atomi utilizzando il software NAMD sviluppato presso l'Università dell'Illinois a Urbana Champaign dal gruppo del defunto biofisico Klaus Schulten.
Il team di Tezcan ha utilizzato un sistema ridotto di sole quattro proteine collegate tra loro, che può essere piastrellato all'infinito per arrivare al fondo di come il cristallo si apre e si chiude. "Il sistema ridotto ci ha permesso di rendere fattibili per noi questi calcoli, perché ci sono ancora centinaia di migliaia di atomi, anche in questo sistema ridotto, " ha detto Tezcan. Il suo team ha sfruttato le funzionalità specifiche di C98RhuA, come l'utilizzo di una singola coordinata di reazione corrispondente alla sua apertura. "Siamo stati davvero in grado di convalidare questo modello come rappresentativo di ciò che abbiamo osservato nell'esperimento, " ha detto Tezcan.
Le simulazioni molecolari di tutti gli atomi dei reticoli cristallini C98RhuA sono state utilizzate per mappare il panorama dell'energia libera. Questo paesaggio energetico sembra un paesaggio naturale, con valli, montagne, e passi di montagna, ha spiegato il coautore dello studio Francesco Paesani, professore di chimica e biochimica alla UCSD.
"Le valli diventano le configurazioni più stabili dei tuoi assemblaggi proteici, "Paesani ha detto che il sistema molecolare preferisce rispetto a dover spendere energia per superare una montagna. E i passi di montagna indicano la strada da una struttura stabile all'altra.
"Tipicamente, i calcoli energetici gratuiti sono molto costosi e impegnativi perché essenzialmente ciò che stai cercando di fare è campionare tutte le possibili configurazioni di un sistema molecolare che contiene migliaia di atomi. E vuoi sapere quante posizioni possono acquisire questi atomi durante una simulazione. Ci vuole molto tempo e molte risorse del computer, " ha detto Paesani.
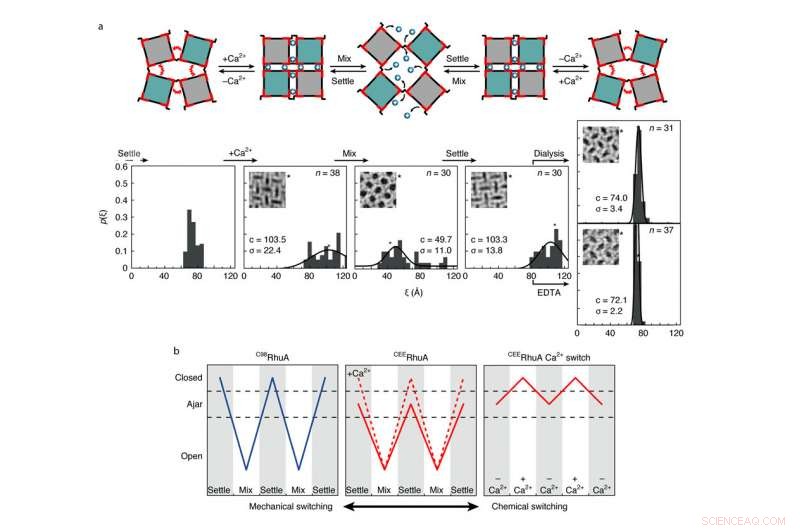
Comportamento di commutazione chimica e meccanica dei cristalli CEERhuA. un, In alto:schema raffigurante tutte le possibili modalità di commutazione dei reticoli CEERhuA. In basso:distribuzione/i sperimentale/i corrispondente/i agli stati direttamente sopra. L'aggiunta di 20 mM Ca2+ alla popolazione di equilibrio 'ajar' di cristalli CEERhuA ha indotto uno spostamento verso conformazioni più chiuse, da cui era possibile la commutazione meccanica simile a C98RhuA. La conformazione socchiusa era completamente recuperabile dopo la rimozione del Ca2+ tramite dialisi o EDTA, fornendo così tre distinte modalità di commutazione. Gli accoppiamenti gaussiani per ciascuna distribuzione sono etichettati con il loro centro (c) e s.d. (σ). n è il numero di cristalli analizzati. La conformazione reticolare di ciascun riquadro è contrassegnata da un asterisco. B, Riepilogo delle modalità di commutazione per i cristalli RhuA. In contrasto con C98RhuA, CEERhuA ha due modalità meccaniche dettate dalla presenza di Ca2+, così come una modalità puramente chimica tramite l'aggiunta o la rimozione di Ca2+. Credito:Robert Alberstein et al.
Per affrontare queste e altre sfide computazionali, Paesani si è aggiudicata le assegnazioni di supercomputer tramite XSEDE, l'ambiente estremo di scoperta della scienza e dell'ingegneria, finanziato dalla National Science Foundation.
"Fortunatamente, XSEDE ci ha fornito un'allocazione su Maverick, i cluster di GPU Computing presso il Texas Advanced Computing Center (TACC), " ha affermato Paesani. Maverick è una risorsa dedicata alla visualizzazione e all'analisi dei dati progettata con 132 unità di elaborazione grafica (GPU) NVIDIA Tesla K40 "Atlas" per la visualizzazione remota e il calcolo GPU per la comunità nazionale.
"Ci è stato molto utile, perché il software NAMD che usiamo funziona molto bene sulle GPU. Questo ci permette di velocizzare i calcoli per ordini di grandezza, " Disse Paesani. "Oggi, possiamo permetterci calcoli che dieci anni fa non potevamo nemmeno sognarci a causa di questi sviluppi, sia sul software NAMD che sull'hardware. Tutti questi cluster di calcolo forniti da XSEDE sono in realtà abbastanza utili per tutte le simulazioni dinamiche molecolari".
Attraverso XSEDE, Paesani utilizzava diversi sistemi di supercalcolo, compreso Gordon, Cometa, e cavalletti al San Diego Supercomputer Center; Kraken presso l'Istituto Nazionale di Scienze Computazionali; e Ranger, precipitoso, e Stampede2 al TACC.
"Poiché tutte le simulazioni sono state eseguite su GPU, Maverick è stata la scelta perfetta per questo tipo di applicazione, " ha detto Paesani.
Calcolo ed esperimento hanno lavorato insieme per produrre risultati. "Penso che questo sia un bellissimo esempio della sinergia tra teoria ed esperimento, Paesani ha detto. "L'esperimento ha posto la prima domanda. La teoria e la simulazione al computer hanno affrontato questa domanda, fornendo una certa comprensione del meccanismo. E poi abbiamo usato la simulazione al computer per fare previsioni e chiedere agli esperimenti di testare la validità di queste ipotesi. Tutto ha funzionato molto bene perché le simulazioni spiegavano gli esperimenti all'inizio. Le previsioni fatte sono state confermate dagli esperimenti alla fine. È un esempio della perfetta sinergia tra esperimenti e modelli teorici".
Tezcan ha aggiunto che "i chimici tradizionalmente amano costruire molecole complesse da blocchi di costruzione più semplici, e si può immaginare di fare una tale combinazione di design, esperimento e calcolo per molecole più piccole per prevedere il loro comportamento. Ma il fatto che possiamo farlo su molecole composte da centinaia di migliaia di atomi non ha precedenti".
Il team scientifico ha anche utilizzato simulazioni di dinamica molecolare per studiare rigorosamente il ruolo dell'acqua nella direzione del movimento reticolare di C98RhuA. "Questo studio ci ha mostrato quanto sia importante il ruolo attivo dell'acqua nel controllo delle dinamiche strutturali di macromolecole complesse, che in biochimica può essere trascurato, " ha detto Tezcan. "Ma questo studio ha mostrato, molto chiaramente, che la dinamica di queste proteine è guidata attivamente dalla dinamica dell'acqua, che penso porti in primo piano l'importanza dell'acqua."
Rob Albertstein, studente laureato nel gruppo Tezcan e primo autore dell'articolo Nature Chemistry, ha aggiunto "Al centro di questa ricerca c'è la comprensione di come le proprietà dei materiali derivano dalla struttura molecolare o atomica sottostante. È molto difficile da descrivere. In questo caso abbiamo davvero cercato di tracciare quella connessione nel modo più chiaro possibile per capirla noi stessi e davvero mostra non solo come dall'esperimento, dove possiamo osservare il comportamento su macroscala di questi materiali, ma poi con il calcolo ricollega quel comportamento a ciò che sta effettivamente accadendo su scala molecolare. Mentre continuiamo a svilupparci come società, dobbiamo sviluppare nuovi materiali per nuovi tipi di problemi globali (purificazione dell'acqua, eccetera), quindi comprendere questa relazione tra la struttura atomica e la proprietà materiale stessa e la capacità di prevederle diventerà sempre più importante".