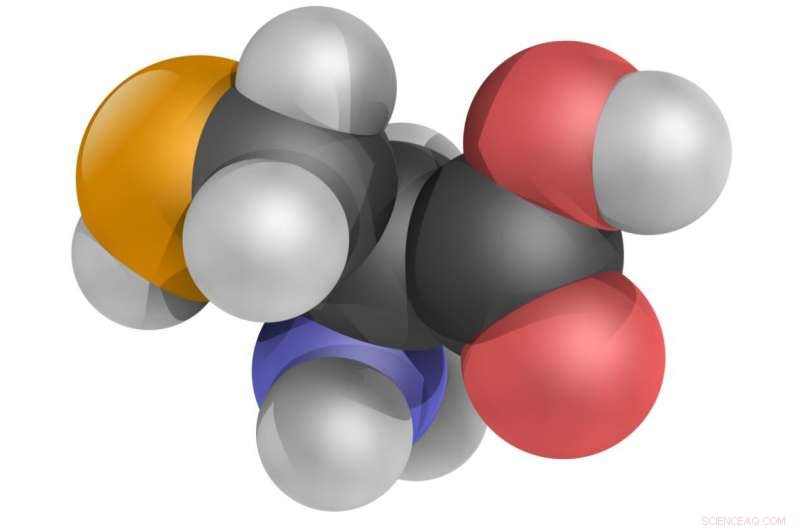
L'aminoacido selenocisteina, Modello di palline 3D. Credito:YassineMrabet/CC BY 3.0/Wikipedia
Quasi ogni processo biologico fondamentale necessario per la vita è svolto dalle proteine. Creano e mantengono le forme di cellule e tessuti; costituiscono gli enzimi che catalizzano le reazioni chimiche che sostengono la vita; agire come fabbriche molecolari, trasportatori e motori; fungere sia da segnale che da ricevitore per le comunicazioni cellulari; e altro ancora.
Composto da lunghe catene di amminoacidi, le proteine svolgono questa miriade di compiti ripiegandosi in precise strutture 3D che governano il modo in cui interagiscono con altre molecole. Poiché la forma di una proteina determina la sua funzione e l'entità della sua disfunzione nella malattia, gli sforzi per illuminare le strutture proteiche sono centrali per tutta la biologia molecolare e, in particolare, scienza terapeutica e lo sviluppo di farmaci salvavita e che alterano la vita.
Negli ultimi anni, i metodi computazionali hanno fatto passi da gigante nel predire come le proteine si ripiegano sulla base della conoscenza della loro sequenza di amminoacidi. Se pienamente realizzato, questi metodi hanno il potenziale per trasformare praticamente tutti gli aspetti della ricerca biomedica. Approcci attuali, però, sono limitati nella scala e nella portata delle proteine che possono essere determinate.
Ora, uno scienziato della Harvard Medical School ha utilizzato una forma di intelligenza artificiale nota come deep learning per prevedere la struttura 3D di qualsiasi proteina in base alla sua sequenza di amminoacidi.
Segnalazione online in Sistemi cellulari il 17 aprile il biologo dei sistemi Mohammed AlQuraishi descrive in dettaglio un nuovo approccio per la determinazione computazionale della struttura proteica, ottenendo un'accuratezza paragonabile agli attuali metodi all'avanguardia ma a velocità fino a un milione di volte più veloci.
"Il ripiegamento delle proteine è stato uno dei problemi più importanti per i biochimici nell'ultimo mezzo secolo, e questo approccio rappresenta un modo fondamentalmente nuovo di affrontare questa sfida, " disse Al Quraishi, docente di biologia dei sistemi presso l'Istituto Blavatnik presso HMS e membro del Laboratorio di Farmacologia dei Sistemi. "Ora abbiamo una prospettiva completamente nuova da cui esplorare il ripiegamento delle proteine, e penso che abbiamo appena iniziato a grattare la superficie".
Facile da affermare
Pur avendo un grande successo, i processi che utilizzano strumenti fisici per identificare le strutture proteiche sono costosi e richiedono tempo, anche con tecniche moderne come la microscopia crioelettronica. Come tale, la stragrande maggioranza delle strutture proteiche e gli effetti delle mutazioni che causano malattie su queste strutture sono ancora in gran parte sconosciuti.
I metodi computazionali che calcolano il modo in cui le proteine si ripiegano hanno il potenziale per ridurre drasticamente il costo e il tempo necessari per determinare la struttura. Ma il problema è difficile e rimane irrisolto dopo quasi quattro decenni di intensi sforzi.
Le proteine sono costruite da una libreria di 20 diversi amminoacidi. Questi si comportano come lettere in un alfabeto, combinando in parole, frasi e paragrafi per produrre un numero astronomico di testi possibili. A differenza delle lettere dell'alfabeto, però, gli amminoacidi sono oggetti fisici posizionati nello spazio 3-D. Spesso, le sezioni di una proteina saranno in stretta vicinanza fisica ma saranno separate da grandi distanze in termini di sequenza, poiché le sue catene di amminoacidi formano anelli, spirali, fogli e torsioni.
"La cosa interessante del problema è che è abbastanza facile da dire:prendi una sequenza e scopri la forma, " Ha detto AlQuraishi. "Una proteina inizia come una stringa non strutturata che deve assumere una forma 3D, e i possibili insiemi di forme in cui può piegarsi una corda sono enormi. Molte proteine sono lunghe migliaia di amminoacidi, e la complessità supera rapidamente la capacità dell'intuizione umana o anche dei computer più potenti."
Difficile da risolvere
Per affrontare questa sfida, gli scienziati sfruttano il fatto che gli amminoacidi interagiscono tra loro in base alle leggi della fisica, ricercare stati energeticamente favorevoli come una palla che rotola in discesa per posarsi in fondo a una valle.
Gli algoritmi più avanzati calcolano la struttura delle proteine eseguendo su supercomputer, o potenza di calcolo di crowdsourcing nel caso di progetti come Rosetta@Home e Folding@Home, per simulare la complessa fisica delle interazioni degli amminoacidi attraverso la forza bruta. Per ridurre gli enormi requisiti di calcolo, questi progetti si basano sulla mappatura di nuove sequenze su modelli predefiniti, che sono strutture proteiche precedentemente determinate attraverso l'esperimento.
Altri progetti come AlphaFold di Google hanno generato un enorme entusiasmo recente utilizzando i progressi dell'intelligenza artificiale per prevedere la struttura di una proteina. Fare così, questi approcci analizzano enormi volumi di dati genomici, che contengono il progetto per le sequenze proteiche. Cercano sequenze in molte specie che probabilmente si sono evolute insieme, utilizzando tali sequenze come indicatori di stretta vicinanza fisica per guidare l'assemblaggio della struttura.
Questi approcci di intelligenza artificiale, però, non predire strutture basate esclusivamente sulla sequenza di amminoacidi di una proteina. Così, hanno un'efficacia limitata per le proteine per le quali non esiste una conoscenza preliminare, proteine uniche evolutive o nuove proteine progettate dall'uomo.
Allenarsi profondamente
Per sviluppare un nuovo approccio, AlQuraishi ha applicato il cosiddetto deep learning differenziabile end-to-end. Questo ramo dell'intelligenza artificiale ha ridotto drasticamente la potenza di calcolo e il tempo necessari per risolvere problemi come il riconoscimento di immagini e parlato, abilitando applicazioni come Siri di Apple e Google Translate.
In sostanza, L'apprendimento differenziabile implica un singolo, enorme funzione matematica - una versione molto più sofisticata di un'equazione di calcolo del liceo - organizzata come una rete neurale, con ogni componente della rete che alimenta le informazioni in avanti e indietro.
Questa funzione può sintonizzarsi e regolarsi, più e più volte a livelli di complessità inimmaginabili, per "imparare" precisamente come una sequenza proteica si relaziona matematicamente alla sua struttura.
AlQuraishi ha sviluppato un modello di deep learning, definita rete geometrica ricorrente, che si concentra sulle caratteristiche chiave del ripiegamento delle proteine. Ma prima che possa fare nuove previsioni, deve essere addestrato utilizzando sequenze e strutture precedentemente determinate.
Per ogni amminoacido, il modello prevede l'angolo più probabile dei legami chimici che collegano l'amminoacido con i suoi vicini. Predice anche l'angolo di rotazione attorno a questi legami, che influenza il modo in cui qualsiasi sezione locale di una proteina è geometricamente correlata all'intera struttura.
Questo viene fatto ripetutamente, con ogni calcolo informato e raffinato dalle posizioni relative di ogni altro amminoacido. Una volta completata l'intera struttura, il modello verifica l'accuratezza della sua previsione confrontandola con la struttura della "verità fondamentale" della proteina.
L'intero processo viene ripetuto per migliaia di proteine note, con l'apprendimento del modello e migliorando la sua accuratezza ad ogni iterazione.
Nuova vista
Una volta addestrato il suo modello, AlQuraishi ha testato il suo potere predittivo. Ha confrontato le sue prestazioni con altri metodi di diversi anni recenti del Critical Assessment of Protein Structure Prediction, un esperimento annuale che testa i metodi computazionali per la loro capacità di fare previsioni utilizzando strutture proteiche che sono state determinate ma non rilasciate pubblicamente.
Ha scoperto che il nuovo modello ha superato tutti gli altri metodi nel prevedere le strutture proteiche per le quali non esistono modelli preesistenti, compresi i metodi che utilizzano dati co-evolutivi. Ha anche superato tutti i metodi tranne i migliori quando erano disponibili modelli preesistenti per fare previsioni.
Sebbene questi guadagni di precisione siano relativamente piccoli, AlQuraishi osserva che eventuali miglioramenti nella fascia alta di questi test sono difficili da ottenere. E poiché questo metodo rappresenta un approccio completamente nuovo al ripiegamento delle proteine, può integrare i metodi esistenti, sia computazionale che fisico, determinare una gamma di strutture molto più ampia di quanto fosse possibile in precedenza.
Sorprendentemente, il nuovo modello esegue le sue previsioni a circa sei-sette ordini di grandezza più velocemente rispetto ai metodi di calcolo esistenti. L'addestramento del modello può richiedere mesi, ma una volta addestrato può fare previsioni in millisecondi rispetto alle ore o ai giorni necessari utilizzando altri approcci. Questo drammatico miglioramento è in parte dovuto alla singola funzione matematica su cui si basa, che richiedono solo poche migliaia di righe di codice per l'esecuzione invece di milioni.
La rapida velocità delle previsioni di questo modello consente nuove applicazioni che prima erano lente o difficili da realizzare, Al Quraishi ha detto, come predire come le proteine cambiano la loro forma mentre interagiscono con altre molecole.
"Approcci di apprendimento profondo, non solo mio, continueranno a crescere nel loro potere predittivo e in popolarità, perché rappresentano un minimo, paradigma semplice in grado di integrare nuove idee più facilmente rispetto agli attuali modelli complessi, " Ha aggiunto.
Il nuovo modello non è subito pronto per l'uso in, dire, scoperta o progettazione di farmaci, Al Quraishi ha detto, perché la sua precisione attualmente si aggira intorno ai 6 angstrom, ancora a una certa distanza dagli 1 o 2 angstrom necessari per risolvere l'intera struttura atomica di una proteina. Ma ci sono molte opportunità per ottimizzare l'approccio, Egli ha detto, comprese ulteriori regole integrative tratte dalla chimica e dalla fisica.
"Prevedere in modo accurato ed efficiente il ripiegamento delle proteine è stato un santo graal per il campo, ed è mia speranza e aspettativa che questo approccio, combinato con tutti gli altri notevoli metodi che sono stati sviluppati, potrà farlo in un prossimo futuro, " AlQuraishi ha detto. "Potremmo risolverlo presto, e penso che nessuno lo avrebbe detto cinque anni fa. È molto eccitante e anche un po' scioccante allo stesso tempo".
Per aiutare gli altri a partecipare allo sviluppo del metodo, AlQuraishi ha reso il suo software e i risultati disponibili gratuitamente tramite la piattaforma di condivisione del software GitHub.
"Una caratteristica notevole del lavoro di AlQuraishi è che un singolo ricercatore, integrato nel ricco ecosistema di ricerca della Harvard Medical School e della comunità biomedica di Boston, può competere con aziende come Google in una delle aree più calde dell'informatica, " ha detto Peter Sorger, HMS Otto Krayer Professore di Farmacologia dei Sistemi presso il Blavatnik Institute presso HMS, direttore del Laboratorio di Farmacologia dei Sistemi presso HMS e mentore accademico di AlQuraishi.
"Non è saggio sottovalutare l'impatto dirompente di persone brillanti come AlQuraishi che lavorano con software open source di pubblico dominio, " ha detto Sorger.