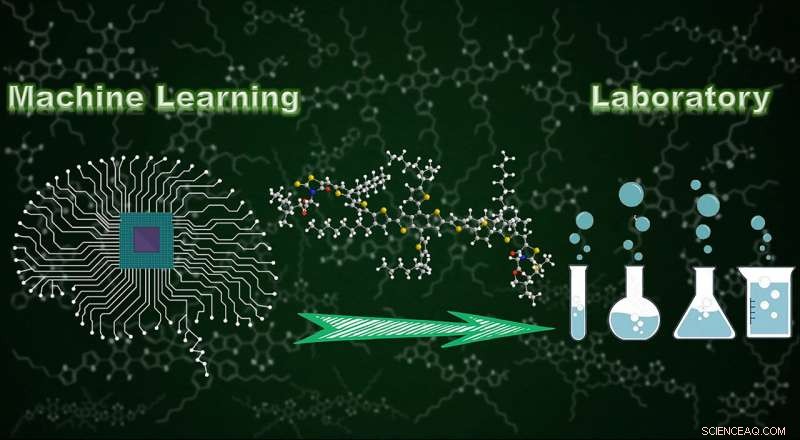
Utilizzo dell'apprendimento automatico per supportare la progettazione molecolare. Credito:Wenbo Sun, Progressi scientifici, doi:10.1126/sciadv.aay4275
Per sintetizzare materiali ad alte prestazioni per il fotovoltaico organico (OPV) che convertono la radiazione solare in corrente continua, gli scienziati dei materiali devono stabilire in modo significativo la relazione tra le strutture chimiche e le loro proprietà fotovoltaiche. In un nuovo studio su Progressi scientifici , Wenbo Sun e un team di ricercatori della School of Energy and Power Engineering, Scuola di Automazione, Informatica, Ingegneria Elettrica e Tecnologia Verde e Intelligente, istituito un nuovo database di più di 1, 700 materiali di donatori utilizzando i rapporti della letteratura esistente. Hanno utilizzato l'apprendimento supervisionato con modelli di apprendimento automatico per costruire relazioni struttura-proprietà e materiali OPV a schermo rapido utilizzando una varietà di input per diversi algoritmi ML.
Utilizzando impronte molecolari (codificando una struttura di una molecola in bit binari) oltre una lunghezza di 1000 bit Sun et al. ottenuto un'elevata precisione di previsione ML. Hanno verificato l'affidabilità dell'approccio esaminando 10 materiali di donatori di nuova concezione per la coerenza tra le previsioni del modello e i risultati sperimentali. I risultati ML hanno presentato un potente strumento per preselezionare nuovi materiali OPV e accelerare lo sviluppo di OPV nell'ingegneria dei materiali.
Le celle fotovoltaiche organiche (OPV) possono facilitare la trasformazione diretta ed economica dell'energia solare in elettricità con una rapida crescita recente per superare i tassi di efficienza di conversione di potenza (PCE). La ricerca tradizionale sull'OPV si è concentrata sulla costruzione di una relazione tra le nuove strutture molecolari dell'OPV e le loro proprietà fotovoltaiche. Il processo tradizionale prevede tipicamente la progettazione e la sintesi di materiali fotovoltaici per l'assemblaggio/ottimizzazione di celle fotovoltaiche. Tali approcci comportano cicli di ricerca dispendiosi in termini di tempo che richiedono un controllo delicato della sintesi chimica e della fabbricazione del dispositivo, fasi sperimentali e purificazione. L'attuale processo di sviluppo dell'OPV è lento e inefficiente con meno di 2000 molecole donatrici di OPV sintetizzate e testate finora. Però, i dati raccolti da decenni di ricerca non hanno prezzo, con potenziali valori ancora da esplorare completamente per generare materiali OPV ad alte prestazioni.
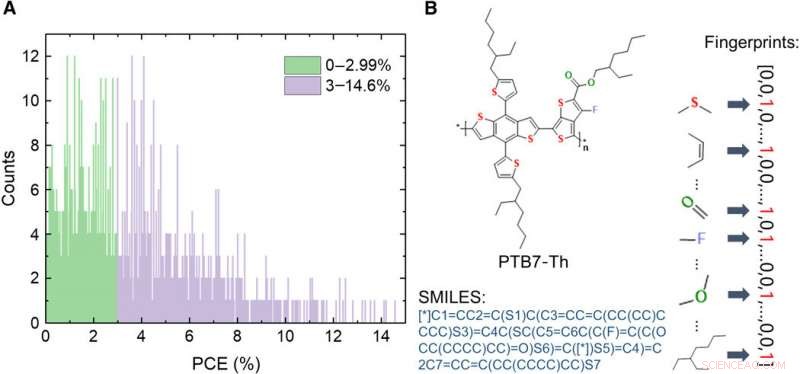
Informazioni sul database dei materiali dei donatori OPV. (A) Distribuzione dei valori PCE delle 1719 molecole nel database. (B) Schemi di espressione di una molecola, compresa l'immagine, sistema di ingresso di linea a input molecolare semplificato (SMILES), e impronte digitali. Credito:progressi scientifici, doi:10.1126/sciadv.aay4275
Per estrarre informazioni utili dai dati, Sole et al. richiedeva un programma sofisticato per eseguire la scansione di un ampio set di dati ed estrarre le relazioni tra le funzionalità. Poiché l'apprendimento automatico (ML) fornisce strumenti computazionali per apprendere e riconoscere modelli e relazioni utilizzando un set di dati di addestramento, il team ha utilizzato un approccio basato sui dati per abilitare il machine learning e prevedere diverse proprietà dei materiali. L'algoritmo ML non doveva comprendere la chimica o la fisica dietro le proprietà dei materiali per svolgere i compiti. Metodi simili hanno recentemente previsto con successo l'attività/proprietà dei materiali durante la scoperta dei materiali, sviluppo di farmaci e progettazione dei materiali. Prima delle applicazioni ML, gli scienziati avevano generato chemininformatica per creare una cassetta degli attrezzi utile.
Gli scienziati dei materiali hanno esplorato solo di recente le applicazioni del ML nel campo dell'OPV. Nel presente lavoro, Sole et al. ha stabilito un database contenente 1719 materiali OPV di donatori testati sperimentalmente raccolti dalla letteratura. Hanno studiato l'importanza dell'espressione del linguaggio di programmazione delle molecole prima di comprendere le prestazioni del machine learning. Hanno quindi testato diversi tipi di espressioni tra cui immagini, stringhe ASCII, due tipi di descrittori e sette tipi di impronte molecolari. Hanno osservato che le previsioni del modello erano in buon accordo con i risultati sperimentali. Gli scienziati si aspettano che il nuovo approccio acceleri notevolmente lo sviluppo di nuovi materiali semiconduttori organici altamente efficienti per applicazioni di ricerca OPV.
Il team di ricerca ha prima trasformato i dati grezzi in una rappresentazione leggibile dalla macchina. Esiste una varietà di espressioni per la stessa molecola che comprende informazioni chimiche molto diverse presentate a diversi livelli astratti. Utilizzando un insieme di modelli ML, Sole et al. hanno esplorato diverse espressioni di una molecola confrontando la loro accuratezza prevista per l'efficienza di conversione di potenza (PCE) per ottenere un'accuratezza del modello di deep learning del 69,41%. Le prestazioni relativamente insoddisfacenti erano dovute alle ridotte dimensioni del database. Ad esempio, in precedenza quando lo stesso gruppo utilizzava un numero maggiore di molecole fino a 50, 000, l'accuratezza del modello di deep learning ha superato il 90 percento. Per addestrare completamente un modello di deep learning, i ricercatori devono implementare un database più ampio contenente milioni di campioni.
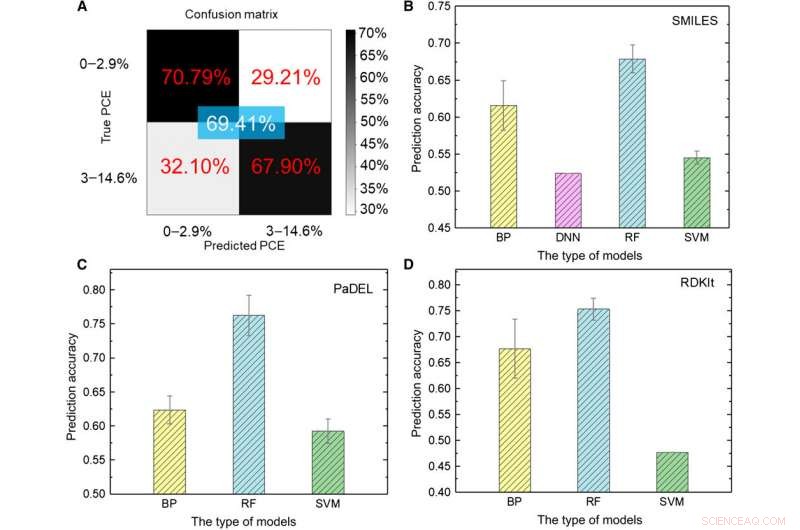
Risultati dei test sui modelli ML. (A) Test del modello di deep learning utilizzando immagini come input. (B a D) Test dei risultati di diversi modelli ML utilizzando (B) SMILES, (C) PALLA, e (D) descrittori RDKIt come input. Credito:progressi scientifici, doi:10.1126/sciadv.aay4275
Sole et al. aveva solo centinaia di molecole in ogni categoria al momento, rendendo difficile per il modello estrarre informazioni sufficienti per una maggiore precisione. Sebbene sia possibile mettere a punto un modello pre-addestrato per ridurre la quantità di dati richiesti, migliaia di campioni sono ancora necessari per realizzare un numero sufficiente di funzioni. Ciò ha portato alla possibilità di aumentare le dimensioni del database quando si utilizzano le immagini per esprimere le molecole.
Gli scienziati hanno utilizzato cinque tipi di algoritmi ML supervisionati nello studio, inclusa (1) rete neurale a propagazione posteriore (BP) (BPNN), (2) rete neurale profonda (DNN), (3) apprendimento profondo, (4) supporta la macchina vettoriale (SVM) e (5) la foresta casuale (RF). Questi erano algoritmi avanzati, dove BPNN, DNN e deep learning erano basati sulla rete neutra artificiale (ANN). Il codice SMILES (sistema di immissione della linea di input molecolare semplificato) ha fornito un'altra espressione originale di una molecola, quale Sun et al. utilizzati come ingressi per quattro modelli. In base ai risultati, la massima precisione si è avvicinata al 67,84 percento per il modello RF. Come prima, a differenza dell'apprendimento profondo, i quattro metodi classici non potevano estrarre caratteristiche nascoste. Nel complesso, SMILES ha ottenuto risultati peggiori delle immagini come descrittori di molecole per prevedere la classe PCE (efficienza di conversione di potenza) nei dati.
I ricercatori hanno quindi utilizzato descrittori molecolari in grado di descrivere le proprietà di una molecola utilizzando una serie di numeri invece dell'espressione diretta di una struttura chimica. Il team di ricerca ha utilizzato due tipi di descrittori PaDEL e RDKIt nello studio. Dopo analisi approfondite su tutti i modelli ML, una grande dimensione dei dati implicava più descrittori irrilevanti per PCE che influenzavano le prestazioni della ANN. Comparativamente, una piccola dimensione dei dati implicava informazioni chimiche inefficienti per addestrare efficacemente i modelli ML, quando si utilizzano descrittori molecolari come input negli approcci ML, la chiave si basava sulla ricerca di descrittori appropriati direttamente correlati all'oggetto di destinazione.
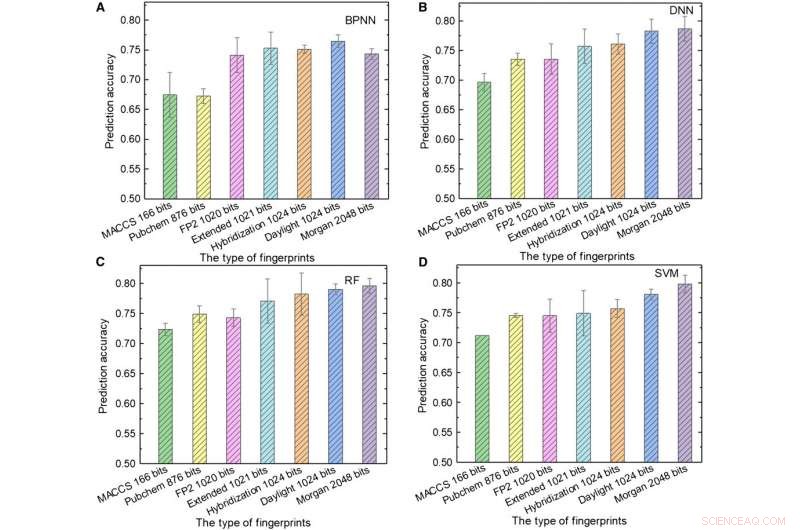
Prestazioni dei modelli ML. (da A a D) I risultati dei test di (A) BPNN, (B) NN, (C) radiofrequenza, e (D) SVM utilizzando diversi tipi di impronte digitali come input. Credito:progressi scientifici, doi:10.1126/sciadv.aay4275.
Il team ha poi utilizzato le impronte digitali molecolari; tipicamente progettato per rappresentare le molecole come oggetti matematici e originariamente creato per identificare gli isomeri. Durante lo screening di database su larga scala, il concetto è rappresentato come un array di bit contenenti "1" se "0" s per descrivere la presenza o l'assenza di sottostrutture o modelli specifici all'interno delle molecole. Sole et al. ha utilizzato sette tipi di impronte digitali come input per addestrare i modelli ML e ha considerato l'influenza della lunghezza dell'impronta digitale sulle prestazioni di previsione di diversi modelli per ottenere impronte digitali diverse. Ad esempio, le impronte digitali del sistema di accesso molecolare (MACCS) contenevano 166 bit ed erano l'input più breve ei risultati erano insoddisfacenti a causa delle loro informazioni limitate.
Sole et al. ha mostrato la migliore combinazione di linguaggio di programmazione e algoritmo ML ottenuta utilizzando impronte digitali di ibridazione di 1024 bit e RF, ottenere una precisione di previsione dell'81,76 percento; dove le impronte digitali di ibridazione rappresentavano gli stati di ibridazione SP2 delle molecole. Quando la lunghezza dell'impronta digitale è aumentata da 166 a 1024 bit, le prestazioni di tutti i modelli ML sono migliorate poiché le impronte digitali più lunghe includevano più informazioni chimiche.
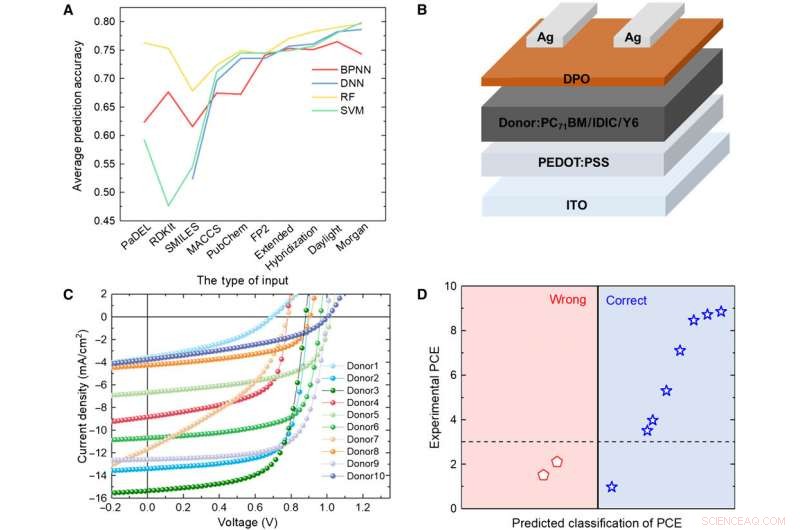
Verifica di modelli ML con esperimento. (A) Confronto dei risultati di quattro diversi modelli. (B) Diagramma schematico dell'architettura cellulare utilizzata in questo studio. (C) Curva J-V della cella solare con lo strato attivo utilizzando il materiale donatore previsto. (D) Risultati della previsione rispetto ai dati sperimentali per i materiali del donatore previsti con l'algoritmo RF e le impronte digitali Daylight. Credito:progressi scientifici, doi:10.1126/sciadv.aay4275.
Per testare l'affidabilità dei modelli ML, Sole et al. sintetizzato 10 nuove molecole donatrici di OPV. Quindi ha utilizzato tre impronte digitali rappresentative per esprimere la struttura chimica delle nuove molecole e ha confrontato i risultati previsti dal modello RF e i valori PCE sperimentali. Il sistema ha classificato otto delle 10 molecole. I risultati hanno indicato il potenziale dei materiali sintetici per le applicazioni OPV con un'ulteriore ottimizzazione sperimentale per due dei nuovi materiali. Un piccolo cambiamento nella struttura potrebbe causare una grande differenza nei valori PCE. incoraggiante, i modelli ML hanno identificato tali modifiche minori per facilitare risultati di previsione favorevoli.
In questo modo, Wenbo Sun e colleghi hanno utilizzato un database della letteratura sui materiali dei donatori OPV e una varietà di espressioni del linguaggio di programmazione (immagini, stringhe ASCII, descrittori e impronte molecolari) per costruire modelli ML e prevedere la corrispondente classe PCE OPV. Il team ha dimostrato uno schema per progettare materiali per donatori di OPV utilizzando approcci ML e analisi sperimentali. Hanno preselezionato un gran numero di materiali dei donatori utilizzando il modello ML per identificare i principali candidati per la sintesi e ulteriori esperimenti. Il nuovo lavoro può accelerare la progettazione di nuovi materiali donatori per accelerare lo sviluppo di OPV PCE elevati. L'uso del machine learning in combinazione con gli esperimenti farà progredire la scoperta dei materiali.
© 2019 Scienza X Rete