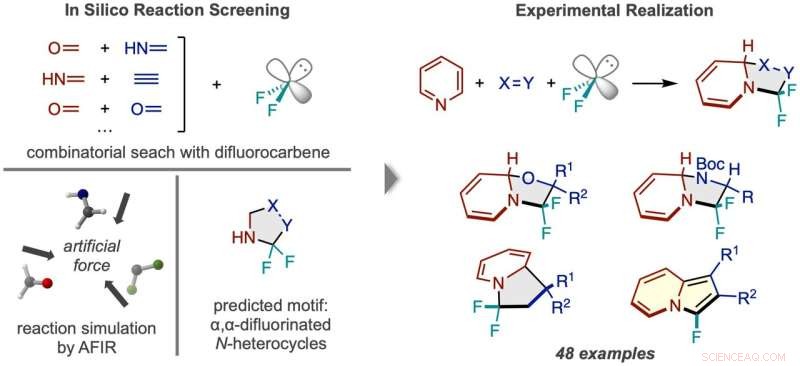
Flusso di lavoro della scoperta delle reazioni tramite screening in silico. (A sinistra) Sono state simulate le reazioni tra difluorocarbene e numerose coppie di piccole molecole, prevedendo un prodotto N-eterociclo fluorurato due volte al carbonio alfa. (A destra) Il quadro di reazione di successo che utilizza la piridina ed esempi dei tipi di composti del prodotto ottenuti. Credito:Sintesi della natura (2022). DOI:10.1038/s44160-022-00128-y
Le simulazioni al computer sono spesso utilizzate come guida in modo che i chimici possano elaborare in modo più efficiente i dettagli esatti di un'idea di reazione generale che hanno in mente, proprio come una bussola aiuta a guidare un esploratore in modo efficiente verso una destinazione sulla loro mappa. Tuttavia, i ricercatori dell'ICReDD hanno fatto un grande passo avanti e hanno utilizzato simulazioni per produrre l'idea generale di una reazione del tutto inimmaginabile, utilizzando efficacemente i calcoli per creare la mappa stessa. Utilizzando il principio di progettazione suggerito dai risultati computazionali, il team ha raggiunto il Motherlode in laboratorio, sviluppando con successo una suite di 48 reazioni che producono composti potenzialmente utili per lo sviluppo di nuovi farmaci.
La presenza e la posizione del fluoro in una molecola influenza spesso l'attività farmacologica di una molecola. I ricercatori dell'ICReDD hanno utilizzato calcoli chimici quantistici per scoprire una reazione che aggiunge selettivamente due atomi di fluoro a una posizione di difficile accesso su un N-eterociclo:molecole con una struttura ad anello di carbonio in cui almeno un carbonio nell'anello viene sostituito con azoto . La capacità di attaccare atomi di fluoro al "carbonio alfa" precedentemente di difficile accesso, il carbonio immediatamente accanto all'azoto nella struttura ad anello, potrebbe portare allo sviluppo di una serie di nuovi farmaci.
Prima di condurre esperimenti in laboratorio, i ricercatori hanno lanciato un'ampia rete, testando computazionalmente la fattibilità di numerose reazioni a 3 componenti utilizzando il metodo della reazione indotta da forza artificiale (AFIR). Hanno simulato la reazione di una molecola di difluorocarbene, che agisce alla fonte degli atomi di fluoro, con varie coppie di piccole molecole caratterizzate da un doppio o triplo legame. Queste simulazioni hanno mostrato che un certo numero di reazioni di formazione dell'anello dovrebbero essere praticabili.
I ricercatori hanno provato una delle reazioni promettenti suggerite dai risultati di calcolo iniziali, ma non hanno avuto successo. A more narrowly focused, optimized computation of the transition state energy of the reaction in question showed that the difluorocarbene molecule more easily reacted with itself than with the desired starting molecules, signaling that an undesired side reaction was likely occurring. This result inspired researchers to change one of the starting materials to the cyclic molecule pyridine, which they anticipated would be able to compete with the unwanted side reaction. This change resulted in the successful synthesis of the desired N-heterocyclic product with two fluorines attached at the alpha carbon position.
The reaction developed here is also significant because it breaks the aromatic system of electrons in the pyridine molecule, a transformation that is especially difficult due to the high stability of aromatic systems. Additionally, the 3-component reaction framework was applied successfully in the lab to a wide range of starting materials, resulting in many new molecules with unique alpha position fluorine substitutions. The large scope of reactivity greatly increases the potential utility of this reaction framework in new drug development.
The researchers see their streamlined screening method as a way to broaden the scope of their search and discover new horizons in chemical reaction design.
"Our study's highlight is the successful demonstration of an in silico reaction screening strategy for reaction development. The computational reaction simulation suggested less-explored three-component reactions of difluorocarbene and two unsaturated molecules, which we successfully realized in experiments," explained lead author Hiroki Hayashi.
"I think the AFIR method is a powerful tool for dictating new research directions in reaction discovery, and we plan to continue building a computation-based reaction development platform by integrating the computational and informatics techniques of ICReDD."
The study was published in Nature Synthesis . + Esplora ulteriormente