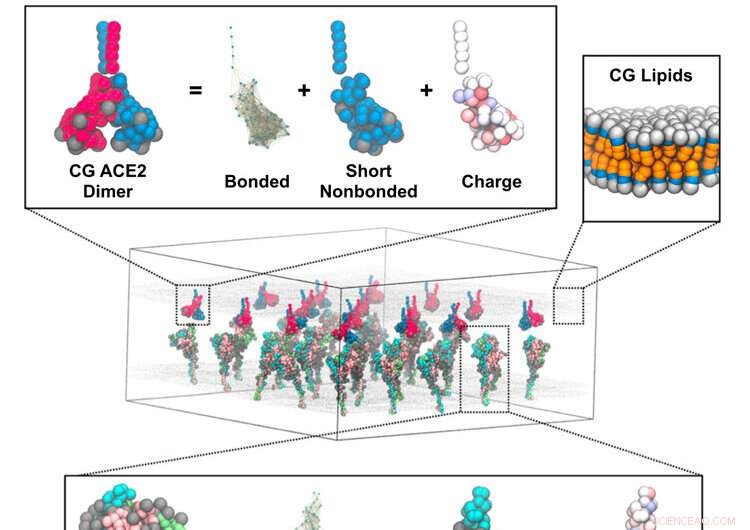
Il meccanismo mediante il quale il coronavirus si fonde con le cellule ospiti è stato suggerito attraverso simulazioni da ricercatori dell'Università di Chicago che utilizzano il supercomputer Frontera al TACC. Rappresentazione rappresentativa di una simulazione a grana fine (CG) di trimeri di punta nella membrana che interagiscono con una membrana adiacente con dimeri ACE2. Gli inserti raffigurano i componenti del modello CG per lo spike trimero (in basso), il dimero ACE2 (in alto a sinistra) e la membrana lipidica (in alto a destra). Credito:Pak, AJ, Yu, A., Ke, Z. et al.
Il mistero di come il virus SARS-CoV-2 infetti le cellule polmonari umane rimane in gran parte nascosto agli scienziati sperimentali. Ora, tuttavia, i dettagli diabolici del meccanismo mediante il quale il coronavirus si fonde con le cellule ospiti sono stati suggeriti attraverso simulazioni dai ricercatori dell'Università di Chicago che utilizzano il supercomputer Frontera presso il Texas Advanced Computing Center (TACC).
I modelli computerizzati mostrano il comportamento cooperativo delle proteine del recettore della cellula ospite che porta alla loro stessa infezione. Il lavoro può essere applicato per aiutare a comprendere la maggiore virulenza delle varianti del coronavirus come delta, omicron e altro.
"Abbiamo scoperto che la proteina spike interagisce con due recettori ACE2 in modo molto cooperativo", ha affermato Gregory Voth, un illustre professore di chimica all'Università di Chicago. "Questa è un'intuizione biofisica fondamentale".
Voth è autore senior dello studio che ha modellato le interazioni tra coronavirus e cellule recettoriali con simulazioni al computer pubblicate sulla rivista Nature Communications nel febbraio 2022.
Come un pallone da calcio con punte, le proteine della punta adornano la superficie del coronavirus. Le punte cercano e si fondono con i recettori della proteina dell'enzima di conversione dell'angiotensina 2 (ACE2) nelle cellule polmonari umane. La proteina spike è composta da due parti principali. Il dominio S1 contiene il dominio di legame del recettore che riconosce le proteine ACE2. E il dominio S2 contiene il macchinario di fusione, che è protetto e coperto come un guscio dal dominio S1.
Le simulazioni rivelano come una proteina del recettore ACE2 trattiene il picco del coronavirus e lo indebolisce mentre l'altra inizia a separarlo. Il dominio S1 quindi si sfalda ed espone il macchinario di fusione. Questo pugno "uno-due" prepara il virus alla fusione e all'ingresso nelle cellule ospiti del polmone umano.
"Sembra che varianti come delta e omicron possano accentuare ulteriormente questo comportamento:è un passaggio chiave. In definitiva, i futuri anticorpi e possibilmente i farmaci molecolari dovrebbero essere in grado di interferire con questo processo", ha affermato Voth.
Voth e colleghi hanno sviluppato quelli che chiamano "modelli dal basso verso l'alto a grana grossa" che hanno preso i dati della tomografia crioelettronica dal laboratorio del coautore dello studio John Briggs del Max Planck Institute of Biochemistry. Lo hanno combinato con simulazioni atomistiche di dinamica molecolare. I dati generati sono stati inseriti in un quadro teorico che ha sviluppato i modelli a grana grossa.
"I modelli a grana grossa sono fino a 1.000 volte più veloci delle simulazioni di dinamica molecolare atomistica diretta, ma mantengono le caratteristiche fisiche essenziali", ha affermato Voth. Questo metodo fornisce un enorme risparmio di tempo e denaro sui calcoli.
Il team scientifico ha ricevuto risorse e servizi di supercomputer dal Consorzio HPC COVID-19, uno sforzo pubblico-privato a sostegno della ricerca COVID-19. Attraverso il consorzio, hanno utilizzato il sistema Frontera finanziato dalla National Science Foundation presso il TACC; il cluster di computer Witherspoon presso IBM Research; e le risorse dell'Oak Ridge Leadership Computing Facility presso l'Oak Ridge National Laboratory.
"Abbiamo calcolato i dati di dinamica molecolare di tutti gli atomi su Frontera e utilizzato gli strumenti di analisi disponibili da TACC:entrambi sono stati molto preziosi", ha affermato Voth.
Il team di Voth ha presentato il suo articolo prima che fossero note le varianti delta e omicron e quindi non ha previsto le mutazioni. Ma sono tornati indietro e hanno rivisto i modelli per indagare sulle varianti.
"Delta ha qualcosa come un'apertura nel picco che si verifica più facilmente rispetto alle precedenti mutazioni del coronavirus", ha detto Voth. "È stato emozionante da un punto di vista scientifico vedere comportamenti che non erano stati visti prima".
Voth ha fatto riferimento a dati di laboratorio di microscopia crioelettronica che mostrano la struttura di una proteina spike solubile con due recettori ACE2 legati ad essa. Ma ha distinto questo esempio cristallizzato da quello che ha studiato usando simulazioni nell'ambiente più realistico di molte proteine che interagiscono tra loro su fogli di membrana.
"I supercomputer, se usati bene e basati su una buona fisica, possono fornire un modo completamente nuovo di guardare questi processi. Attraverso la simulazione al computer, si possono studiare cose che al momento non possono essere fatte con gli esperimenti. La simulazione e gli esperimenti funzionano molto bene insieme, mano nella mano", ha detto Voth. + Esplora ulteriormente