La sintesi o lo studio di determinati materiali in un ambiente di laboratorio spesso pone sfide dovute a problemi di sicurezza, condizioni sperimentali poco pratiche o vincoli di costo. In risposta, gli scienziati si rivolgono sempre più a metodi di deep learning che implicano lo sviluppo e l'addestramento di modelli di machine learning per riconoscere modelli e relazioni nei dati che includono informazioni su proprietà, composizioni e comportamenti dei materiali.
Utilizzando il deep learning, gli scienziati possono fare rapidamente previsioni sulle proprietà dei materiali in base alla composizione, alla struttura e ad altre caratteristiche rilevanti, identificare potenziali candidati per ulteriori indagini e ottimizzare le condizioni di sintesi.
Ora, in uno studio apparso sull'International Union of Crystallography Journal (IUCrJ) , il professor Takashiro Akitsu, il professore assistente Daisuke Nakane e il signor Yuji Takiguchi dell'Università delle Scienze di Tokyo (TUS) hanno utilizzato l'apprendimento profondo per prevedere magneti a singola molecola (SMM) da un pool di 20.000 complessi metallici. Questa strategia innovativa semplifica il processo di scoperta dei materiali riducendo al minimo la necessità di lunghi esperimenti.
I magneti a singola molecola (SMM) sono complessi metallici che dimostrano un comportamento di rilassamento magnetico a livello di singola molecola, dove i momenti magnetici subiscono cambiamenti o rilassamento nel tempo. Questi materiali hanno potenziali applicazioni nello sviluppo di memoria ad alta densità, dispositivi spintronici molecolari quantistici e dispositivi di calcolo quantistico. Gli SMM sono caratterizzati dall'avere un'elevata barriera energetica effettiva (Ueff ) affinché il momento magnetico si inverta. Tuttavia, questi valori sono generalmente compresi tra decine e centinaia di Kelvin, il che rende difficile sintetizzare gli SMM.
I ricercatori hanno utilizzato il deep learning per identificare la relazione tra strutture molecolari e comportamento delle SMM nei complessi metallici con ligandi di tipo salen. Questi complessi metallici sono stati scelti perché possono essere facilmente sintetizzati complessando aldeidi e ammine con vari metalli 3d e 4f.
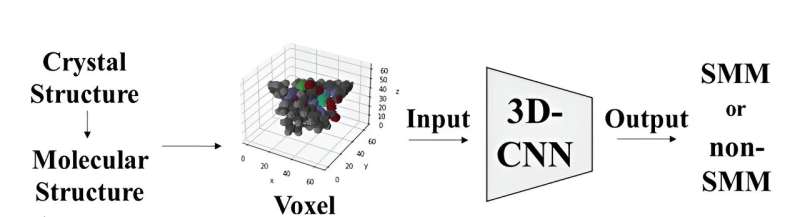
Per il set di dati, i ricercatori hanno lavorato a lungo per esaminare 800 articoli dal 2011 al 2021, raccogliendo informazioni sulla struttura cristallina e determinando se questi complessi mostravano un comportamento SMM. Inoltre, hanno ottenuto dettagli strutturali 3D delle molecole dal database strutturale di Cambridge.
La struttura molecolare dei complessi è stata rappresentata utilizzando voxel o pixel 3D, dove a ciascun elemento è stato assegnato un valore RGB univoco. Successivamente, queste rappresentazioni di voxel sono servite come input per un modello di rete neurale convoluzionale 3D basato sull'architettura ResNet. Questo modello è stato specificamente progettato per classificare le molecole come SMM o non SMM analizzando le loro immagini molecolari 3D.
Quando il modello è stato addestrato su un set di dati di strutture cristalline di complessi metallici contenenti complessi di tipo salen, ha raggiunto un tasso di precisione del 70% nella distinzione tra le due categorie. Quando il modello è stato testato su 20.000 strutture cristalline di complessi metallici contenenti basi di Schiff, ha scoperto con successo i complessi metallici segnalati come magneti a molecola singola.
"Questo è il primo rapporto sull'apprendimento profondo sulle strutture molecolari degli SMM", afferma il prof. Akitsu.
Molte delle strutture SMM previste coinvolgevano complessi multinucleari di disprosio, noti per il loro elevato Ueff valori. Sebbene questo metodo semplifichi il processo di scoperta dell’SMM, è importante notare che le previsioni del modello si basano esclusivamente su dati di addestramento e non collegano esplicitamente le strutture chimiche con i relativi calcoli chimici quantistici, un metodo preferito nella progettazione molecolare assistita dall’intelligenza artificiale. Sono necessarie ulteriori ricerche sperimentali per ottenere dati sul comportamento delle SMM in condizioni uniformi.
Tuttavia, questo approccio semplificato presenta i suoi vantaggi. Riduce la necessità di calcoli computazionali complessi ed evita il compito impegnativo di simulare il magnetismo.
Il Prof. Akitsu conclude:"L'adozione di un simile approccio può guidare la progettazione di molecole innovative, determinando risparmi significativi in termini di tempo, risorse e costi nello sviluppo di materiali funzionali."
Ulteriori informazioni: Yuji Takiguchi et al, La previsione delle proprietà dei magneti di una singola molecola tramite l'apprendimento profondo, IUCrJ (2024). DOI:10.1107/S2052252524000770
Fornito dall'Università delle Scienze di Tokyo