Gli scienziati IBM hanno sviluppato un nuovo approccio per simulare le molecole su un computer quantistico che potrebbe un giorno aiutare a rivoluzionare la chimica e la scienza dei materiali. Gli scienziati hanno utilizzato con successo sei qubit su un processore quantistico a sette qubit appositamente costruito per affrontare il problema della struttura molecolare dell'idruro di berillio (BeH2), la più grande molecola simulata fino ad oggi su un computer quantistico. I risultati dimostrano un percorso di esplorazione per i sistemi quantistici a breve termine per migliorare la nostra comprensione di complesse reazioni chimiche che potrebbero portare ad applicazioni pratiche. Credito:Kandala et al.; Natura
La simulazione di molecole su computer quantistici è diventata molto più semplice con l'hardware quantistico superconduttore di IBM. In un recente articolo di ricerca pubblicato su Natura , Eigensolver quantistico variazionale efficiente dall'hardware per piccole molecole e magneti quantistici, implementiamo un nuovo algoritmo quantistico in grado di calcolare in modo efficiente lo stato energetico più basso di piccole molecole. Mappando la struttura elettronica degli orbitali molecolari su un sottoinsieme del nostro processore quantistico a sette qubit appositamente costruito, abbiamo studiato molecole precedentemente inesplorate con i computer quantistici, compreso l'idruro di litio (LiH) e l'idruro di berillio (BeH2). La particolare codifica da orbitali a qubit studiata in questo lavoro può essere utilizzata per semplificare le simulazioni di molecole ancora più grandi e ci aspettiamo l'opportunità di esplorare tali simulazioni più grandi in futuro, quando la potenza di calcolo quantistica (o "volume quantistico") dei sistemi IBM Q è aumentata.
Mentre BeH2 è la più grande molecola mai simulata da un computer quantistico fino ad oggi, il modello considerato della molecola stessa è ancora abbastanza semplice da simulare esattamente con i computer classici. Questo lo ha reso un banco di prova per spingere i limiti di ciò che il nostro processore a sette qubit potrebbe raggiungere, approfondire la nostra comprensione dei requisiti per migliorare l'accuratezza delle nostre simulazioni quantistiche, e gettare gli elementi fondamentali necessari per esplorare tali studi sull'energia molecolare.
Le migliori simulazioni di molecole oggi vengono eseguite su computer classici che utilizzano metodi approssimativi complessi per stimare l'energia più bassa di un hamiltoniano molecolare. Un "Hamiltoniano" è un operatore energetico della meccanica quantistica che descrive le interazioni tra tutti gli orbitali elettronici e i nuclei degli atomi costituenti. Lo stato di "energia più bassa" dell'Hamiltoniana molecolare determina la struttura della molecola e come interagirà con altre molecole. Tali informazioni sono fondamentali per i chimici per progettare nuove molecole, reazioni, e processi chimici per applicazioni industriali.
Qubit:orbitale
Sebbene il nostro processore quantistico a sette qubit non sia completamente corretto dagli errori e tollerante ai guasti, i tempi di coerenza dei singoli qubit durano circa 50 µs. È quindi molto importante utilizzare un algoritmo quantistico molto efficiente per ottenere il massimo dalla nostra preziosa coerenza quantistica e cercare di comprendere le strutture molecolari. L'algoritmo deve essere efficiente in termini di numero di qubit utilizzati e numero di operazioni quantistiche eseguite.

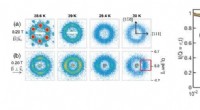
Applicazione alla chimica quantistica. AC, Risultati sperimentali (cerchi neri pieni), superfici energetiche esatte (linee tratteggiate) e grafici di densità (ombreggiatura; vedere scale di colori) dei risultati delle simulazioni numeriche, per diverse distanze interatomiche per H2 (a), LiH (b) e BeH2 (c). I risultati sperimentali e numerici presentati sono per circuiti di profondità d = 1 Le barre di errore sui dati sperimentali sono più piccole delle dimensioni dei marker. I grafici di densità sono ottenuti da 100 risultati numerici a ciascuna distanza interatomica. I riquadri superiori in ogni pannello evidenziano i qubit utilizzati per l'esperimento e le porte di risonanza incrociata (frecce, etichettato CRc–t; dove "c" indica il qubit di controllo e "t" il qubit di destinazione) che costituiscono UENT. I riquadri inferiori sono rappresentazioni della geometria molecolare (non in scala). Per tutte e tre le molecole, la deviazione dei risultati sperimentali dalle curve esatte è ben spiegata dalle simulazioni stocastiche. Credito:Kandala et al.; Natura
Il nostro schema contrasta con gli algoritmi di simulazione quantistica studiati in precedenza, che si concentrano sull'adattamento dei classici schemi di simulazione molecolare all'hardware quantistico, senza tener conto in modo efficace dei costi generali limitati degli attuali dispositivi quantistici realistici.
Quindi, invece di forzare i metodi di calcolo classici sull'hardware quantistico, abbiamo invertito l'approccio e ci siamo chiesti:come possiamo estrarre la massima potenza computazionale quantistica dal nostro processore a sette qubit?
La nostra risposta a questo combina una serie di tecniche efficienti dall'hardware per attaccare il problema:
Con i futuri processori quantistici, che avrà più volume quantico, saremo in grado di esplorare la potenza di questo approccio alla simulazione quantistica per molecole sempre più complesse che vanno oltre le capacità di calcolo classiche. La capacità di simulare accuratamente le reazioni chimiche, è favorevole agli sforzi per scoprire nuovi farmaci, fertilizzanti, anche nuove fonti energetiche sostenibili.
Gli esperimenti che dettagliamo nel nostro documento non sono stati eseguiti sui nostri processori a cinque e 16 qubit attualmente disponibili pubblicamente sul cloud. Ma gli sviluppatori e gli utenti dell'esperienza IBM Q possono ora accedere ai notebook Jupyter di chimica quantistica sul repository github QISKit. Sul sistema a cinque qubit, gli utenti possono esplorare la simulazione dell'energia dello stato fondamentale per le piccole molecole di idrogeno e LiH. Notebook per molecole più grandi sono disponibili per coloro che hanno accesso beta al processore aggiornato a 16 qubit.