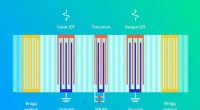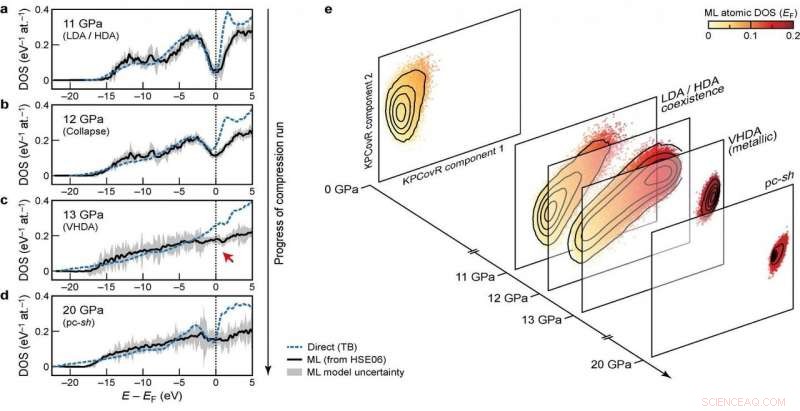
Densità di stati elettronici (DOS) nelle varie fasi della corsa di compressione Credit:@Michele Ceriotti
La combinazione di calcoli di strutture elettroniche e tecniche di apprendimento automatico (ML) è diventato un approccio comune nella modellazione atomistica della materia. L'utilizzo delle due tecniche insieme ha consentito ai ricercatori, ad esempio, per creare modelli che utilizzano le coordinate atomiche come unici input per prevedere in modo economico qualsiasi proprietà che può essere calcolata dai calcoli dei principi primi che erano stati utilizzati per addestrarli.
Mentre i primi e ormai più avanzati sforzi si sono concentrati sull'utilizzo di previsioni di energie totali e forze atomiche per costruire potenziali interatomici, sforzi più recenti hanno preso di mira proprietà aggiuntive di cristalli e molecole come energie di ionizzazione, schermature chimiche NMR, proprietà di risposta dielettrica e densità di carica. Nel documento "Apprendimento della densità elettronica degli stati nella materia condensata, "Ceriotti e colleghi si concentrano sulla densità elettronica degli stati (DOS), un'altra quantità che sta alla base di molte proprietà utili dei materiali, alcuni dei quali possono essere osservati sperimentalmente.
Il DOS è essenzialmente il numero di diversi stati che gli elettroni possono occupare ad un particolare livello di energia, e può essere utilizzato, ad esempio, calcolare il contributo elettronico alla capacità termica nei metalli e la densità dei portatori di carica libera nei semiconduttori. È un proxy indiretto per proprietà come l'energy band gap, l'energia di banda e lo spettro di assorbimento ottico.
"Prevedere il DOS è un esercizio interessante in sé perché è essenzialmente la descrizione più semplice possibile della struttura elettronica oltre l'immagine dello stato fondamentale, " ha detto Ceriotti. "È utile anche perché ci sono molte proprietà che si possono calcolare partendo dal DOS, rendendolo un ottimo esempio di come la prossima generazione di modelli ML possa essere utilizzata in modo simile ai calcoli di strutture elettroniche, utilizzandole in modo indiretto per calcolare quantità intermedie che possono poi essere facilmente elaborate per valutare proprietà più difficili da apprendere direttamente".
Nello sviluppo del modello, il gruppo ha cercato di assicurare la trasferibilità attraverso le diverse fasi, nonché la scalabilità a sistemi di grandi dimensioni. Il loro approccio finale, che esamina come le diverse configurazioni atomiche influenzano la distribuzione dei livelli di energia, soddisfa questi obiettivi:è stato in grado di apprendere e prevedere il DOS calcolato da DFT per un set di dati diversificato di strutture di silicio, coprendo un'ampia gamma di condizioni termodinamiche e diverse fasi. Inoltre scala linearmente, piuttosto che con il cubo del numero di atomi come con i calcoli della struttura elettronica, rendendolo applicabile a grandi strutture. Finalmente, il modello ha consentito un'analisi del DOS locale, dando ai ricercatori la possibilità di esaminare l'interazione tra motivi strutturali e struttura elettronica.
La combinazione di trasferibilità, e scalabilità delle previsioni a sistemi di grandi dimensioni, rendere il modello applicabile per affrontare questioni aperte di lunga data nella scienza dei materiali. Il nuovo framework è già stato utilizzato per chiarire le proprietà elettroniche di una simulazione di 100.000 atomi di silicio amorfo, subendo una serie di transizioni di fase quando compresso a 20 Gpa, in un articolo pubblicato su Natura oggi in collaborazione con un team composto da ricercatori di Oxford, Cambridge, il Laboratorio di Ricerca Navale degli Stati Uniti e l'Università dell'Ohio. Il DOS previsto viene utilizzato anche per spiegare come le trasformazioni strutturali indotte dalla pressione si accoppiano alla struttura elettronica del materiale.
La combinazione del nuovo modello con uno dei modelli di energia potenziale ben consolidati consente inoltre di calcolare i contributi elettronici a proprietà macroscopiche come la capacità termica dei metalli e di eseguire simulazioni che tengono conto degli effetti della temperatura elettronica finita, come dimostrato in un altro articolo di prossima pubblicazione che discute le proprietà del nichel alle alte temperature. Infatti, il nuovo modello è un passo fondamentale verso l'obiettivo di MARVEL di sviluppare modelli di apprendimento automatico integrati che aumentano, e forse eventualmente sostituiscono, i costosi calcoli di strutture elettroniche.
"Ci sono altre proprietà oltre alla densità elettronica degli stati, come eccitazioni ottiche, e risposta NMR, che siamo stati in grado di prevedere con precisione con l'apprendimento automatico". Ceriotti ha detto. "Se possiamo usarli tutti in combinazione con potenziali interatomici economici e precisi ci permetterà di descrivere tutte le proprietà dei materiali con la stessa accuratezza ottenuta con l'elettronica calcolo della struttura, ma ad una piccola frazione del costo."