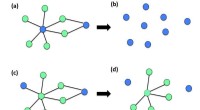
Rappresentazione schematica dei diversi termini energetici che contribuiscono all'energia di adsorbimento, e differenza di densità di carica di 2H-P dopo l'adsorbimento su Cu(111) a 12,8 Angstrom di separazione. Credito:M. Müller/TU Monaco di Baviera
Mentre continuiamo a ridurre i componenti elettronici, i metodi di produzione top-down iniziano ad avvicinarsi a un limite fisico su scala nanometrica. Piuttosto che continuare a intaccare questo limite, una soluzione interessante prevede l'utilizzo dell'autoassemblaggio dal basso verso l'alto di blocchi molecolari per costruire dispositivi su scala nanometrica.
L'autoassemblaggio di successo è una danza elaboratamente coreografata, in cui le forze attrattive e repulsive all'interno delle molecole, tra ogni molecola e le sue vicine, e tra le molecole e la superficie che le sostiene, bisogna tener conto di tutti. Per comprendere meglio il processo di autoassemblaggio, ricercatori del Politecnico di Monaco hanno caratterizzato i contributi di tutte le componenti di interazione, come il legame covalente e le interazioni di van der Waals tra le molecole e tra le molecole e una superficie.
"In un caso ideale, il dispositivo più piccolo possibile ha le dimensioni di un singolo atomo o molecola, "ha detto Katharina Diller, che ha lavorato come ricercatore post-dottorato nel gruppo di Karsten Reuter presso l'Università tecnica di Monaco. Reuter e i suoi colleghi presentano il loro lavoro questa settimana in Il Giornale di Fisica Chimica .
Uno di questi esempi è un interruttore a singola porfirina, che occupa una superficie di un solo nanometro quadrato. La molecola di porfina, che è stato oggetto di questo studio, è ancora più piccolo di questo. Le porfirine sono un gruppo di composti chimici ad anello che comprendono in particolare l'eme - responsabile del trasporto di ossigeno e anidride carbonica nel flusso sanguigno - e la clorofilla. Nelle applicazioni di derivazione sintetica, le porfirine sono studiate per i loro potenziali utilizzi come sensori, coloranti fotosensibili nelle celle solari organiche, e magneti molecolari.
I ricercatori della TU Munich hanno valutato le interazioni della molecola di porfirina 2H-porfina utilizzando la teoria del funzionale della densità, un metodo di modellazione computazionale della meccanica quantistica utilizzato per descrivere le proprietà elettroniche di molecole e materiali. Le loro simulazioni sono state eseguite presso il supercomputer ad alte prestazioni SuperMUC a Leibniz-Rechenzentrum a Garching.
I substrati metallici che i ricercatori hanno scelto per l'assemblaggio delle molecole di porfirina, le superfici monocristalline ravvicinate di rame e argento, sono ampiamente utilizzati come substrati nella scienza delle superfici. Ciò è dovuto alla natura densa delle superfici, che consentono alle molecole di esibire un ambiente di adsorbimento regolare. Inoltre, rame e argento reagiscono in modo diverso con le poririne:la molecola si adsorbe più fortemente sul rame, mentre l'argento fa un lavoro migliore nel mantenere intatta la struttura elettronica della molecola, consentendo ai ricercatori di monitorare una varietà di effetti concorrenti per applicazioni future.
Nella loro simulazione, molecole di porfirina sono state poste su una lastra di rame o argento, che è stato ripetuto periodicamente per simulare una superficie estesa. Dopo aver trovato la geometria ottimale in cui le molecole si adsorbirebbero sulla superficie, i ricercatori hanno alterato le dimensioni della lastra metallica per aumentare o diminuire la distanza tra le molecole, simulando così differenti coperture molecolari. La configurazione computazionale ha dato loro un interruttore per attivare e disattivare i contributi energetici delle molecole vicine, per osservare l'interazione delle singole interazioni.
Diller e Reuter, insieme ai colleghi Reinhard Maurer e Moritz Müller, chi è il primo autore sulla carta, hanno scoperto che le deboli interazioni di van der Waals a lungo raggio hanno dato il maggior contributo all'interazione molecola-superficie, e ha mostrato che i metodi spesso impiegati per quantificare le spese elettroniche nel sistema devono essere usati con cautela. Sorprendentemente, mentre le interazioni direttamente tra le molecole sono trascurabili, il ricercatore ha trovato indicazioni per interazioni molecola-molecola mediate dalla superficie a coperture molecolari più elevate.
"L'analisi della struttura elettronica e dei singoli componenti di interazione ci permette di comprendere meglio l'autoassemblaggio del porfino adsorbito su rame e argento, e consente inoltre previsioni per analoghi della porfirina più complessi, " Diller ha detto. "Queste conclusioni, però, venire senza ancora considerare gli effetti del moto atomico a temperatura finita, che non abbiamo studiato in questo lavoro."