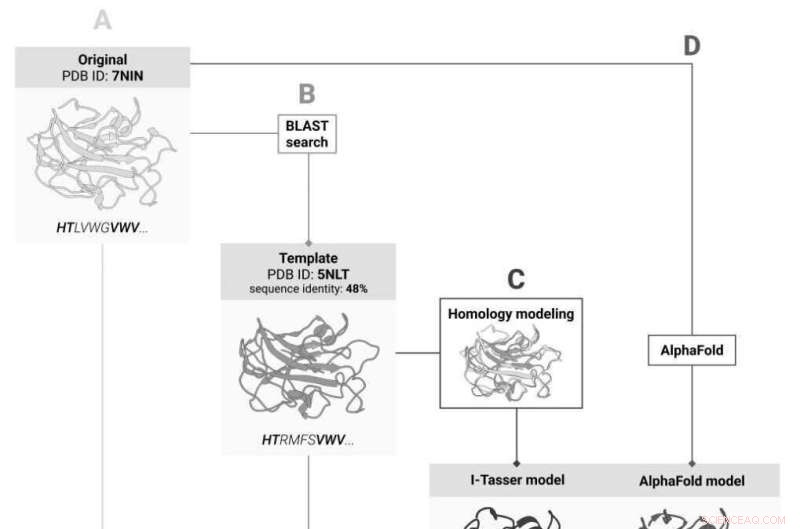
Quattro modi per prevedere i cambiamenti nella stabilità della proteina dopo la mutazione:(A) dalla struttura della proteina originale; (B) dalla struttura del suo omologo; (C) dalla struttura della proteina originale prevista in base alla struttura dell'olomlog e (D) dalla struttura prevista dall'intelligenza artificiale basata sulla sequenza di amminoacidi. Credito:Skolkovo Institute of Science and Technology
I ricercatori dello Skoltech Center for Molecular and Cellular Biology hanno confrontato diversi metodi di predizione della struttura proteica in termini di valutazione della stabilità delle proteine mutanti e hanno ottenuto lo stesso risultato per le strutture previste dall'IA e tridimensionali sperimentali (3D) di proteine con sequenze di amminoacidi simili. Tuttavia, il tentativo di prevedere la struttura della proteina mirata dalla struttura nota del suo "parente" ha solo reso la previsione meno accurata. I risultati del team faciliteranno i calcoli preliminari nella valutazione dei cambiamenti di stabilità causati dalla mutazione. La ricerca è stata pubblicata su Bioinformatica .
Gli esperimenti biologici spesso coinvolgono proteine mutanti, necessarie per lo studio della struttura e delle funzioni delle proteine o dei processi cellulari, nonché per l'ingegneria delle proteine. È noto che le mutazioni influenzano la struttura e la stabilità di una proteina. Poiché gli esperimenti sono troppo costosi e richiedono molto tempo, gli scienziati stanno creando una soluzione alternativa sotto forma di metodi computazionali per valutare l'impatto delle mutazioni sulla stabilità. Tuttavia, le loro applicazioni richiedono la conoscenza della struttura 3D di una proteina.
Una struttura 3D sperimentale non è disponibile per tutte le proteine ed è probabile che manchi per quella presa di mira dal team. Se questo è il caso, i modelli 3D degli omologhi della proteina, cioè i suoi "parenti più stretti", possono fornire l'ancora di salvezza, perché è noto il grado di somiglianza nelle sequenze di amminoacidi che assicura una buona corrispondenza tra le strutture 3D delle proteine. La soluzione sarebbe prima prevedere la struttura della proteina in base alla struttura nota del suo omologo e quindi calcolare l'impatto delle mutazioni per il modello previsto.
Grazie alla svolta dello scorso anno nella previsione della struttura delle proteine, gli scienziati ora hanno un'alternativa:invece di prevedere la struttura 3D basata su omologhi, possono utilizzare lo strumento AlphaFold basato sull'intelligenza artificiale che prevede la struttura della proteina dalla sequenza di amminoacidi e ha già affrontato con la stragrande maggioranza delle proteine conosciute fino ad oggi.
Nel loro recente studio, i ricercatori Skoltech hanno deciso di scoprire quale di questi approcci funziona meglio per prevedere i cambiamenti di stabilità in seguito alla mutazione. Per quanto accurato possa essere AlphaFold, trovare la struttura della proteina attraverso esperimenti rimane ancora il "gold standard". Nel confrontare i due approcci, il team ha utilizzato sette metodi di valutazione della stabilità e ha confrontato i risultati con quelli di AlphaFold e I-Tasser, il miglior sistema di previsione della struttura basato sull'omologo. Inoltre, i ricercatori hanno verificato se possono saltare la previsione della struttura basata sull'omologo e calcolare la stabilità per la struttura nota della proteina omologa.
"Abbiamo deciso di scoprire fino a che punto ci saremmo discostati da una previsione accurata se avessimo utilizzato la struttura proteica "vicina" invece di quella reale. Si è scoperto che la fase di previsione basata sull'omologia peggiora solo le cose producendo un risultato meno accurato. Abbiamo dimostrato che non fa praticamente alcuna differenza se si utilizza la struttura sperimentale dell'omologo o la previsione di AlphaFold.In un certo senso, si trattava di convalida:di fronte a un nuovo metodo, devi prima verificare se funziona per il tuo compito Questo è esattamente quello che abbiamo fatto", primo autore dello studio, Skoltech Ph.D. studentessa Marina Pak, commenti.
"Con tutto questo clamore su AlphaFold, alcuni scienziati e non professionisti ritengono che abbia risolto tutti i problemi di ricerca sulle proteine nella biologia computazionale, ma non è così. Ad esempio, la previsione dei cambiamenti di stabilità indotti dalla mutazione mostra ancora un'affidabilità piuttosto bassa, anche sebbene il cambiamento di stabilità sia tra i fattori chiave della funzionalità delle proteine. Uno strumento che potrebbe determinare in modo inequivocabile l'impatto della mutazione sulla stabilità aiuterebbe sia nella pianificazione dell'esperimento che nell'interpretazione dei risultati. Supponiamo che per una proteina non sia ottimale in termini di stabilità, desideriamo trovare mutazioni che lo rendano stabile nelle condizioni desiderate, ad esempio assicurando che rimanga attivo ad alta temperatura. Una volta che saremo in grado di farlo solo attraverso i calcoli, l'approccio alla riprogettazione e all'ottimizzazione delle proteine cambierà drasticamente ". l'autore principale dello studio, l'assistente professore di Skoltech Dmitry Ivankov conclude.
Sebbene la previsione del cambiamento di stabilità appaia più facile rispetto alla previsione della struttura 3D, rimane comunque una sfida intrattabile anche per l'IA. I dati scarsi di addestramento sono solo uno dei problemi:AlphaFold aveva quasi 200.000 strutture proteiche da addestrare, ma i dati sperimentali sui cambiamenti di stabilità ammontano a migliaia di set mentre coprono solo poche dozzine di proteine uniche. Gli autori sperano che se saranno disponibili più dati e i ricercatori mostreranno un maggiore interesse per l'attività, sarà presto possibile fare una svolta. + Esplora ulteriormente