Quando gli agricoltori del XX secolo osservarono per la prima volta una malattia neurologica inspiegabile nelle pecore, la chiamarono scrapie. Decenni dopo, gli scienziati scoprirono che il colpevole era una proteina mal ripiegata, ora nota come prione, capace di indurre disturbi cerebrali fatali sia negli animali che negli esseri umani.
I prioni sono conformazioni proteiche anomale che possono replicarsi e causare malattie neurodegenerative fatali come il morbo della mucca pazza, il morbo di Creutzfeldt‑Jakob e la malattia del deperimento cronico.
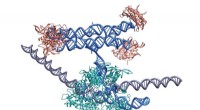
I prioni sono particelle infettive proteiche prive di acidi nucleici. Si formano quando una normale proteina cellulare (spesso chiamata PrP C ) si ripiega in una conformazione che causa la malattia (PrP Sc ). La forma mal ripiegata è ricca di struttura beta-sheet, mentre la proteina sana è prevalentemente ad alfa-elica. Questo cambiamento strutturale consente al prione di modellare il ripiegamento errato di altre proteine normali, provocando una cascata di danni.
A differenza dei virus o dei batteri, i prioni si riproducono senza DNA o RNA. La proteina patogena induce cambiamenti conformazionali nelle proteine sane, convertendole nella forma mal ripiegata. Questo processo autocatalitico consente ai prioni di moltiplicarsi indipendentemente dal materiale genetico, un fenomeno che rimane oggetto di intensa ricerca.
Negli esseri umani, i disturbi da prioni includono:
Negli animali, le malattie da prioni comprendono:
Tutte queste condizioni sono progressive, neurodegenerative e attualmente incurabili. I sintomi tipicamente comportano demenza, disfunzione motoria e infine la morte.
Agenzie di ricerca come CDC e l'OMS mantenere dati aggiornati sulle epidemie da prioni e sui progressi della ricerca.
Sebbene non esistano trattamenti definitivi, gli studi in corso si concentrano sulla rilevazione precoce, sugli anticorpi terapeutici e su nuovi inibitori di piccole molecole che potrebbero interrompere la cascata del misfolding.