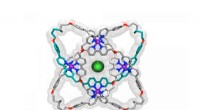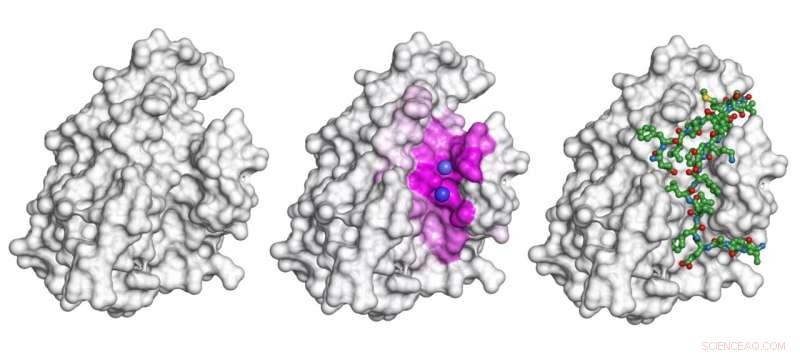
La forma grigia è una proteina. Per lo scenario di questa proteina che si lega al peptide mostrato come un modello verdastro a forma di bastone e palla a destra, il modello presentato nello studio evidenzia la superficie coinvolta nell'interazione (l'area rosa al centro) e prevede gli esatti siti di legame (sfere viola). Credito:Igor Kozlovskii e Petr Popov / Skoltech
Due ricercatori di Skoltech hanno presentato un modello di rete neurale altamente efficiente che utilizza i dati sulla struttura delle proteine per prevedere quali delle loro parti interagiscono con altre molecole biologiche chiamate peptidi. Sapere questo è utile per sviluppare farmaci a base di peptidi, che possono influenzare le interazioni proteina-proteina all'interno delle cellule in modo mirato e non tossico, regolazione di un'ampia gamma di processi cellulari. Lo studio è uscito nel Journal of Chemical Information and Modeling .
Le proteine sono il macchinario delle cellule, in movimento, impegnarsi l'uno con l'altro, ed eseguire ogni tipo di operazione. I farmacologi sono sempre stati incuriositi dalla prospettiva di armeggiare con le interazioni tra proteine. Eppure sembravano essere off-limits come potenziali bersagli farmacologici:le molecole terapeutiche più grandi, chiamati biologici, non poteva penetrare nella cellula per agire sulle proteine, mentre gli agenti a piccole molecole spesso si sono rivelati incapaci di tale azione.
peptidi, che mediano o regolano naturalmente circa il 40% dei processi cellulari, occupano una promettente via di mezzo e offrono prospettive per i farmaci mirati alle interazioni proteina-proteina. I peptidi offrono il meglio di entrambi i mondi:come le piccole molecole, possono penetrare nella membrana cellulare per raggiungere effettivamente i loro obiettivi, e mostrano anche una bassa tossicità, insieme a un'elevata affinità e specificità (azione forte e mirata), i segni distintivi dei farmaci biologici.
Per progettare farmaci a base di peptidi, i farmacologi devono conoscere i cosiddetti siti di legame per ogni dato bersaglio proteico. Questo è, le macchie sulla proteina che possono legarsi a un peptide. Più tali siti sono conosciuti, maggiori sono le opportunità per la progettazione di farmaci.
I ricercatori possono identificare sperimentalmente i siti di legame, Per esempio, utilizzando la cristallografia a raggi X, che rivela la struttura 3D delle proteine cristallizzate studiando come diffrange i raggi X. Ma questo è molto costoso da fare per un lungo elenco di molecole, e i metodi computazionali offrono un'alternativa più rapida ed economica. Alcuni di loro si basano su tecniche di apprendimento automatico, e man mano che si accumulano più dati sulle strutture dei complessi proteina-peptide, questi metodi diventano più potenti e forniscono previsioni del sito di legame sempre migliori.
Nel loro articolo del 22 luglio sul Journal of Chemical Information and Modeling , Skoltech Ph.D. lo studente Igor Kozlovskii e l'assistente professore Petr Popov del gruppo iMolecule hanno presentato un metodo computazionale chiamato BiteNetPp, che sfrutta la potenza delle reti neurali convoluzionali 3D per rilevare i siti di legame proteina-peptide. In BiteNetPp, una struttura proteica nota viene alimentata a una rete neurale, che poi evidenzia siti di legame peptidico sospetti, ed emette una serie di coordinate 3D presunte, insieme ai relativi punteggi di probabilità.
Petr Popov commenta l'approccio al rilevamento del sito di legame come riconoscimento dell'immagine, originariamente introdotto nel precedente articolo del team e riportato nello studio riportato in questa storia:"Proprio come le reti neurali possono essere addestrate a riconoscere, dire, pedoni o ciclisti in normali foto 2D, consideriamo il rilevamento del sito di legame come l'individuazione di un particolare tipo di oggetto in un'immagine. La differenza è che usiamo i dati della struttura atomica 3D come input, quindi il modello opera sui 'voxel, "un analogo tridimensionale dei pixel".
Il modello appena presentato si basa in realtà su quello del documento precedente. "Questo si chiama adattamento del dominio. BiteNetPp è il primo modello ad essere stato messo a punto su un set di dati proteina-peptide dopo essere stato inizialmente addestrato su dati di piccole molecole proteiche, "Spiega Popov. "Si può immaginare questo come un modello di formazione per identificare i luoghi in cui i ciclisti tendono a fermarsi per strada, ma inizi con i dati su dove i pedoni tendono a fermarsi e solo dopo estendi il tuo dominio ai ciclisti. Piuttosto che iniziare da zero, riaddestrare il modello, anticipando che i "siti vincolanti" per i ciclisti potrebbero condividere alcune somiglianze con quelli che attirano i pedoni:sai, bancarelle di gelati, semafori, questo genere di cose."
I creatori del modello hanno dimostrato che BiteNetPp supera costantemente i metodi all'avanguardia esistenti confrontando le loro previsioni per quei siti di legame proteina-peptide noti attraverso osservazioni sperimentali. È importante sottolineare che il nuovo modello impiega meno di un secondo per analizzare una singola struttura proteica, rendendolo adatto per studi su larga scala. Ci sono migliaia di interazioni proteina-proteina potenzialmente mirabili da farmaci a base di peptidi, quindi i metodi computazionali devono essere abbastanza veloci da rendere fattibile il loro screening in un contesto farmacologico.