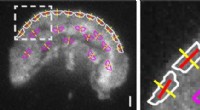
Sbloccare informazioni biologiche da dati genomici complessi di singole cellule è appena diventato più semplice e preciso, grazie all'innovativo strumento scLENS sviluppato dal Biomedical Mathematics Group all'interno del Centro IBS per le scienze matematiche e computazionali guidato dal ricercatore capo Kim Jae Kyoung, che è anche un professore al KAIST. Ciò rappresenta un significativo passo avanti nel campo della trascrittomica unicellulare.
La ricerca è pubblicata sulla rivista Nature Communications .
L'analisi genomica di una singola cellula è una tecnica avanzata che misura l'espressione genica a livello di singola cellula, rivelando cambiamenti e interazioni cellulari che non sono osservabili con i tradizionali metodi di analisi genomica. Se applicata ai tessuti tumorali, questa analisi può delineare la composizione di diversi tipi di cellule all'interno di un tumore, fornendo informazioni su come il cancro progredisce e identificando i geni chiave coinvolti durante ogni fase della progressione.
Nonostante l’immenso potenziale dell’analisi genomica di una singola cellula, gestire l’enorme quantità di dati che genera è sempre stato impegnativo. La quantità di dati copre l’espressione di decine di migliaia di geni in centinaia o migliaia di singole cellule. Ciò non solo si traduce in set di dati di grandi dimensioni, ma introduce anche distorsioni legate al rumore, che derivano in parte dalle attuali limitazioni di misurazione.
L'autore corrispondente Kim Jae Kyoung ha sottolineato:"C'è stato un notevole progresso nelle tecnologie sperimentali per l'analisi dei trascrittomi di singole cellule negli ultimi dieci anni. Tuttavia, a causa delle limitazioni nei metodi di analisi dei dati, c'è stata una lotta per utilizzare appieno i dati preziosi ottenuti attraverso costi e tempi elevati."
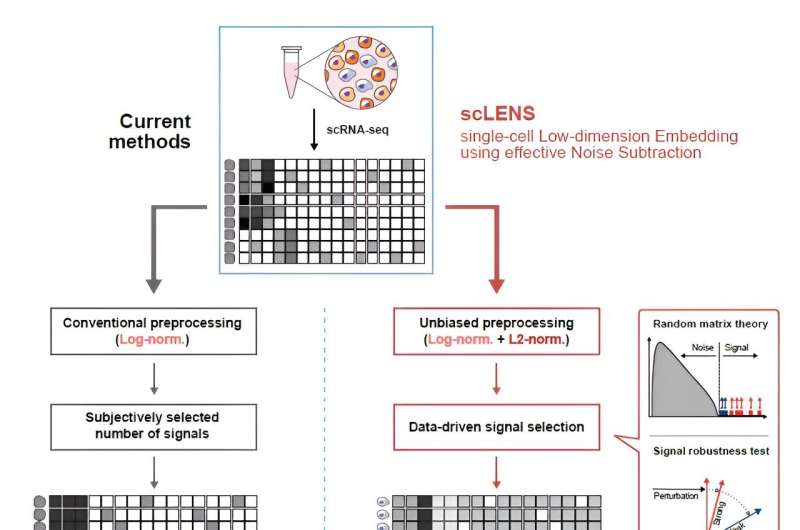
Nel corso degli anni i ricercatori hanno sviluppato numerosi metodi di analisi per distinguere i segnali biologici da questo rumore. Tuttavia, l’accuratezza di questi metodi è stata meno che soddisfacente. Un problema critico è che la determinazione delle soglie di segnale e rumore spesso dipende dalle decisioni soggettive degli utenti.
Lo strumento scLENS di nuova concezione sfrutta la teoria della matrice casuale e il test di robustezza del segnale per differenziare automaticamente i segnali dal rumore senza fare affidamento sull'input soggettivo dell'utente.
Il primo autore Kim Hyun ha dichiarato:"In precedenza, gli utenti dovevano decidere arbitrariamente la soglia per il segnale e il rumore, il che comprometteva la riproducibilità dei risultati dell'analisi e introduceva la soggettività. scLENS elimina questo problema rilevando automaticamente i segnali utilizzando solo la struttura intrinseca dei dati".
Durante lo sviluppo di scLENS, i ricercatori hanno identificato le ragioni fondamentali delle imprecisioni nei metodi di analisi esistenti. Hanno scoperto che i metodi di preelaborazione dei dati comunemente utilizzati distorcono sia i segnali biologici che il rumore. Il nuovo approccio di preelaborazione offerto da scLENS è esente da tali distorsioni.
Risolvendo i problemi relativi alla soglia di rumore determinata dalla scelta soggettiva dell'utente e alla distorsione del segnale nella preelaborazione convenzionale dei dati, scLENS supera significativamente i metodi esistenti in termini di precisione. Inoltre, scLENS automatizza il laborioso processo di selezione della dimensione del segnale, consentendo ai ricercatori di estrarre i segnali biologici in modo comodo e automatico.
Ci Kim ha aggiunto:"scLENS risolve i principali problemi nell'analisi dei dati del trascrittoma di una singola cellula, migliorando sostanzialmente l'accuratezza e l'efficienza durante tutto il processo di analisi. Questo è un ottimo esempio di come le teorie matematiche fondamentali possano guidare l'innovazione nella ricerca nelle scienze della vita, consentendo ai ricercatori di fare di più rispondi in modo rapido e accurato a domande biologiche e scopri segreti della vita precedentemente nascosti."
Ulteriori informazioni: Hyun Kim et al, scLENS:rilevamento del segnale basato sui dati per un'analisi imparziale dei dati scRNA-seq, Nature Communications (2024). DOI:10.1038/s41467-024-47884-3
Informazioni sul giornale: Comunicazioni sulla natura
Fornito da Institute for Basic Science