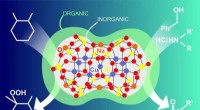
Rappresentazione artistica di molecole organiche adsorbenti su una superficie di silicio. Credito:Aaron Beller
Man mano che si sono resi disponibili nuovi metodi per comprendere e manipolare la materia ai suoi livelli più fondamentali, i ricercatori che lavorano nel campo interdisciplinare della scienza dei materiali hanno avuto sempre più successo nel sintetizzare nuovi tipi di materiali. Spesso l'obiettivo dei ricercatori del settore è progettare materiali che incorporino proprietà che possono essere utili per svolgere funzioni specifiche. Tali materiali possono, Per esempio, essere più chimicamente stabile o resistente alla rottura fisica, hanno caratteristiche elettromagnetiche vantaggiose, o reagire in modi prevedibili a condizioni ambientali specifiche.
Il dottor Ralf Tonner e il suo gruppo di ricerca presso l'Università di Marburg stanno affrontando la sfida di progettare materiali funzionali in un modo insolito, applicando approcci basati sulla chimica computazionale. Utilizzo delle risorse informatiche presso il Centro di calcolo ad alte prestazioni di Stoccarda (HLRS), uno dei tre centri di supercalcolo nazionali tedeschi che compongono il Gauss Center for Supercomputing, Tonner modella i fenomeni che si verificano su scala atomica e subatomica per capire come fattori come la struttura molecolare, proprietà elettroniche, legame chimico, e le interazioni tra gli atomi influenzano il comportamento di un materiale.
"Quando studi come, Per esempio, una molecola si adsorbe su una superficie, "Tonner spiega, "altri scienziati descriveranno spesso quel fenomeno con metodi della fisica, teoria dello stato solido, o strutture a bande. Riteniamo che possa essere molto utile anche chiedere, come guarderebbe un chimico a quello che sta succedendo qui?" Da questo punto di vista, Tonner è interessato a esplorare se la comprensione delle reazioni chimiche, come gli atomi si legano insieme in molecole e reagiscono quando vengono messi in contatto l'uno con l'altro, possa offrire nuove e utili intuizioni.
In una nuova pubblicazione in WIREs Computational Molecular Science , Tonner e la sua collaboratrice Lisa Pecher evidenziano la capacità degli approcci di chimica computazionale che utilizzano il calcolo ad alte prestazioni per rivelare fenomeni interessanti che si verificano tra molecole organiche e superfici. Dimostrano anche più in generale come queste interazioni possono essere comprese rispetto al mondo molecolare e allo stato solido. Le conoscenze acquisite potrebbero essere utili nella progettazione di superfici modellate, un obiettivo degli scienziati che lavorano sulla prossima generazione di più potenti, semiconduttori più efficienti.
Portare il calcolo alla chimica
Gli atomi si legano insieme per formare molecole e composti quando si avvicinano l'uno all'altro e poi scambiano o condividono gli elettroni che orbitano attorno ai loro nuclei. Gli atomi specifici coinvolti, le forme fisiche che assumono le molecole, le loro proprietà energetiche, e il modo in cui interagiscono con altre molecole vicine sono tutte proprietà che conferiscono a un composto le sue proprietà uniche. Tali caratteristiche possono determinare se è probabile che i composti rimangano stabili, o se sollecitazioni come variazioni di temperatura o pressione potrebbero influenzare la loro reattività.
Tonner utilizza un approccio computazionale chiamato teoria del funzionale della densità (DFT) per esplorare tali caratteristiche su scala quantistica; questo è, alla scala in cui la meccanica newtoniana viene sostituita dal mondo molto più strano della meccanica quantistica (a distanze inferiori a 100 nanometri). La DFT utilizza le informazioni sulle variazioni della densità degli elettroni all'interno di una molecola, una quantità che può anche essere misurata sperimentalmente utilizzando una tecnologia ampiamente utilizzata chiamata diffrazione dei raggi X, per ricavare l'energia del sistema. Questo, a sua volta, consente ai ricercatori di dedurre le interazioni tra nuclei e le interazioni tra elettroni e nuclei, fattori critici per la comprensione dei legami e delle reazioni chimiche.
DFT può fornire utili, sebbene statico, informazioni sui profili energetici dei composti che studiano. Per comprendere meglio come si comportano effettivamente i sistemi di molecole quando interagiscono con una superficie, Il gruppo di Tonner utilizza anche il calcolo ad alte prestazioni presso HLRS per eseguire simulazioni di dinamica molecolare. Qui, gli scienziati osservano come il sistema di molecole si sviluppa nel tempo, a livello di atomi ed elettroni e su scale temporali di picosecondi (un picosecondo è un trilionesimo di secondo).
Tali calcoli utilizzano in genere 2, 000-3, 000 core di calcolo, in esecuzione su un problema per una settimana, e Tonner ha previsto un budget di circa 30 milioni di ore CPU presso HLRS per l'attuale ciclo di finanziamento di due anni.

Rappresentazione artistica di molecole organiche adsorbenti su una superficie di silicio. Credito:Aaron Beller
"L'aumento della potenza di calcolo ha permesso alla chimica computazionale e alla chimica quantistica di descrivere sistemi molecolari reali. Solo 15-20 anni fa, le persone potevano guardare solo piccole molecole e dovevano fare approssimazioni piuttosto forti, " Spiega Tonner. "Negli ultimi anni, le comunità di chimica computazionale e teoria dello stato solido hanno risolto il problema di parallelizzare i loro codici per operare in modo efficiente su sistemi di calcolo ad alte prestazioni. Man mano che i supercomputer diventano più grandi, prevediamo di essere in grado di sviluppare modelli sempre più realistici per i sistemi sperimentali nella scienza dei materiali".
Verso i semiconduttori a base di luce
Un'area in cui Tonner sta attualmente utilizzando la chimica computazionale è lo studio di modi per migliorare il silicio da utilizzare in nuovi tipi di semiconduttori. Questo problema ha acquisito urgenza negli ultimi anni, poiché è diventato chiaro che l'industria della microelettronica sta raggiungendo i limiti della sua capacità di migliorare i semiconduttori utilizzando solo il silicio.
Come riportano Tonner e colleghi sperimentali in un recente articolo sul Beilstein Journal of Organic Chemisty, la funzionalizzazione del silicio con composti come il fosfuro di gallio (GaP) o l'arseniuro di gallio (GaAs) potrebbe consentire la progettazione di nuovi tipi di semiconduttori. Questa ricerca, basato in un campo chiamato fotonica del silicio, postula che tali nuovi materiali renderebbero possibile utilizzare la luce invece degli elettroni per il trasporto del segnale, sostenere lo sviluppo di dispositivi elettronici migliorati.
"Per fare questo, "Tonner spiega, "abbiamo davvero bisogno di capire come appaiono e si comportano le interfacce tra il silicio e questi composti organici. La reazione tra queste due classi di materiali deve procedere in modo molto controllato in modo che l'interfaccia sia il più perfetta possibile. Con la chimica computazionale possiamo guardare ai dettagli elementari di queste interazioni e processi."
Per esempio, per ricoprire una lastra di silicio, le molecole precursori liquide per gli atomi costituenti dell'arseniuro di gallio sono poste in un gorgogliatore, dove vengono poi portati in fase gassosa. Queste molecole precursori sono composte dagli atomi necessari per il nuovo materiale (gallio, arsenico) e ioni o molecole chiamate ligandi per stabilizzarli nella fase liquida e gassosa. Questi ligandi vengono successivamente persi nel processo di deposizione e quando il silicio viene inserito nel sistema, le molecole precursori vengono adsorbite sulla superficie solida del silicio. Dopo l'adsorbimento e la perdita dei ligandi, gli atomi di gallio e arseniuro si attaccano al silicio, formare un film di GaAs.
Il modo in cui gli atomi sono disposti quando si adsorbono su una superficie è determinato dal legame chimico. La forza di questi legami e la densità con cui vengono adsorbite le molecole precursori del GaAs è influenzata non solo dalla distanza tra loro e la superficie del silicio ma anche dalle interazioni tra le molecole precursori stesse. In un tipo di interazione, chiamato Pauli repulsione, nuvole di elettroni si sovrappongono e si respingono, facendo diminuire l'energia disponibile per il legame. In un altro, chiamata interazione di dispersione attrattiva, i cambiamenti nelle posizioni elettroniche in un atomo causano la ridistribuzione degli elettroni in altri atomi, portando in armonia i movimenti degli elettroni e abbassando l'energia del sistema totale.
In precedenza, è stato suggerito che le relazioni repulsive tra gli atomi sono il fattore più importante per "orientare" gli atomi al loro posto quando si adsorbono su una superficie. Usando la teoria del funzionale della densità e osservando le caratteristiche intriganti di come gli elettroni sono distribuiti, i ricercatori hanno determinato che la capacità degli atomi di guidare altri atomi in posizione sulla superficie può anche derivare da interazioni dispersive attraenti.
Una migliore comprensione di queste interazioni fondamentali dovrebbe aiutare i progettisti di semiconduttori otticamente attivi a migliorare l'adsorbimento delle molecole precursori sul silicio. Questo, a sua volta, consentirebbe di combinare la conduzione del segnale luminoso con la microelettronica a base di silicio, unendo il meglio dei due mondi nella conduzione ottica ed elettronica.
Per Tonner, l'utilizzo dei metodi dei primi principi in chimica per le applicazioni della scienza dei materiali è molto promettente. "La teoria oggi è molto spesso presa come complemento all'indagine sperimentale, " dice. "Sebbene la sperimentazione sia estremamente importante, il nostro obiettivo finale è che la teoria sia predittiva in modi che ci permettano di fare i primi passi nella progettazione dei materiali ispirata ai primi principi. Lo vedo come un obiettivo a lungo termine".