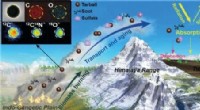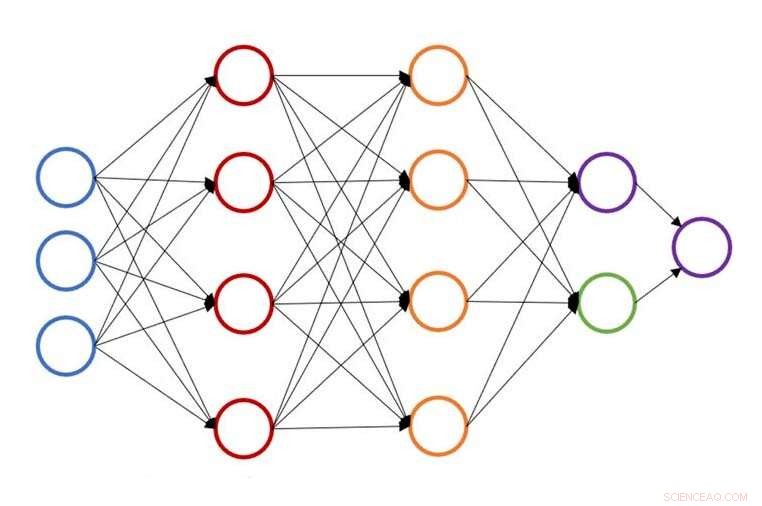
Illustrazione di una singola struttura di rete neurale per la previsione dell'energia atomica. Credito:John Kitchin, Università Carnegie Mellon
Apprendimento automatico, un metodo di analisi dei dati utilizzato per automatizzare la costruzione di modelli analitici, ha rimodellato il modo in cui scienziati e ingegneri conducono la ricerca. Una branca dell'intelligenza artificiale (AI) e dell'informatica, il metodo si basa su un gran numero di algoritmi e ampi set di dati per identificare modelli e prendere importanti decisioni di ricerca.
Le applicazioni delle tecniche di apprendimento automatico stanno emergendo nell'area della catalisi superficiale, consentendo simulazioni più estese di nanoparticelle, studi di segregazione, ottimizzazione della struttura, apprendimento al volo dei campi di forza, e screening ad alto rendimento. Però, lavorare con grandi quantità di dati può spesso essere un compito lungo e dispendioso dal punto di vista computazionale.
Ottimizzazione della geometria, spesso il fattore limitante nelle simulazioni molecolari, è una parte fondamentale dei materiali computazionali e della scienza delle superfici. Consente ai ricercatori di trovare strutture atomiche allo stato fondamentale e percorsi di reazione, proprietà utilizzate per stimare le proprietà cinetiche e termodinamiche delle strutture molecolari e cristalline. Mentre vitale, il processo può essere relativamente lento, richiedendo un gran numero di calcoli da completare.
Alla Carnegie Mellon University, John Kitchin sta lavorando per accelerare questo processo fornendo un metodo di apprendimento attivo basato su rete neurale che accelera l'ottimizzazione geometrica per più configurazioni contemporaneamente. Il nuovo modello riduce il numero di calcoli della teoria del funzionale della densità (DFT) o della teoria del mezzo efficace (EMT) dal 50 al 90 percento, consentendo ai ricercatori di svolgere lo stesso lavoro in meno tempo o più lavori nello stesso lasso di tempo.
"Normalmente, quando eseguiamo l'ottimizzazione della geometria, partiamo da zero, " ha detto Kitchin. "I calcoli raramente traggono beneficio da qualcosa che sapevamo in passato."
"Aggiungendo un modello surrogato al processo, gli abbiamo permesso di fare affidamento su calcoli precedenti, piuttosto che ricominciare da capo ogni volta."
Lo studio illustra l'accelerazione su diversi casi studio, comprese le superfici con adsorbati, superfici metalliche nude, ed elastico spinto per due reazioni. In ogni caso, il pacchetto Python per l'ottimizzazione dell'ambiente di simulazione atomica (ASE) consentiva un numero inferiore di calcoli DFT rispetto al metodo standard.
Il pacchetto Python ASE-optimizer è stato messo a disposizione di colleghi ingegneri e scienziati per rendere più semplice l'utilizzo dell'apprendimento attivo dell'ensemble di reti neurali per l'ottimizzazione della geometria.